Introduction
Imagine you have two complete mtDNA genomes, A and B, and you’d like to determine which one of the two is the ancestor of the other, with no other information whatsoever. This is simply impossible to determine, since they have only a single, mutual relationship with each other, which consists of some number of bases in common. Yes, of course, if you have access to other information about where in history certain genes or other genome regions fall, or other information concerning the provenance of the genomes, then you might be able to actually date the genomes. However, the point is, that in the absence of information exogenous to the two genomes, you simply cannot determine which of the two is the ancestor of the other, given only the raw genome sequences themselves.
Now assume instead you’re given three genomes, A, B, and C, where all three genomes are from distinct populations. Further, assume that it is in fact the case that genome A comes from the ethnic ancestor population of genomes B and C. Note that this does not require genome A to be older than genomes B and C, and in fact, all genomes could have been sourced from living persons. The point is instead that genomes B and C are literally mutations of genome A. It follows that it is far more likely that genomes B and C have less in common with each other, than either of them do with genome A. To assume otherwise implies that genomes B and C developed even more common bases over time simply by chance. It is instead far more likely that mutations over time cause B and C to diverge along separate paths of mutation. For intuition, imagine that two people are tossing independent and unbiassed coins. Further, assume that by chance, they happen to both throw three heads in a row. From that point going forward, they are almost certainly going to diverge along two different paths of outcomes, and as the number of coin tosses increases, the probability of both throwing exactly the same sequence starts to decrease rapidly. Therefore, if we assume that A is the common ancestor of both B and C, then it is most likely that A has more bases in common with both B and C, than B and C have in common with each other.
This is a very simple condition to test for, which reduces to a simple inequality. Specifically, let 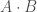



However, this does not tell you how long ago the mutations occurred, and because mtDNA is at times incredibly stable, over thousands of years, you simply can’t claim an amount of time has lapsed that generated the observed mutations. It is instead a simply ordinal relationship, that again does not imply ancestry, but is instead consistent with ancestry. As a consequence, the most narrow interpretation is that all relationships that fail this test almost certainly rule out ancestry. Nonetheless, the results produced are consistent with known history and common sense.
Application to Data
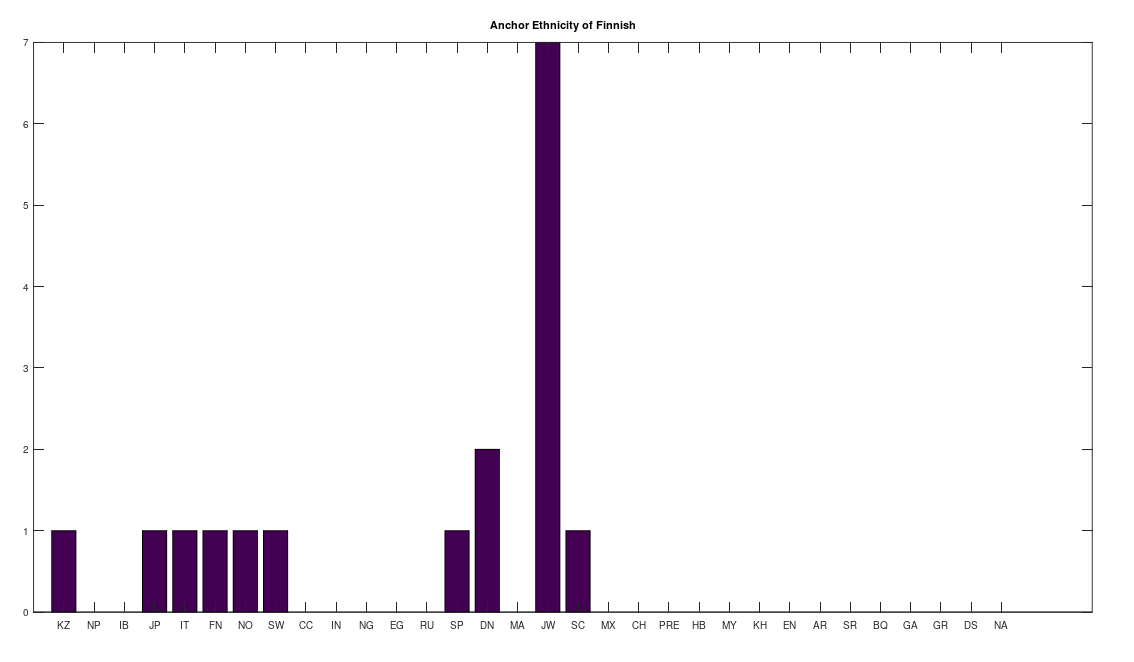
Below is the graph produced by analyzing the Kazakh genomes in the dataset linked to at the end of this article. A given vertex is attached to another pair, if they satisfy the inequality above. The graphs are automatically generated by the attached Octave code below, using SageMath, you just need to copy / paste the SageMath code, which is written to a file. As you can plainly see, the Kazakh people are a cradle of humanity, and in fact, this is consistent with known history, as Homo sapiens have been in Kazakhstan for hundreds of thousands of years. The vertex labels on the graph are the genome row numbers in the dataset, and the color key to the right tells you to which population a given genome belongs.


There are some other examples that superficially seem to go the wrong way in time, but this is incorrect. Specifically, if you run this analysis on the Mexican genomes, you find that one of the Mexican genomes is the ancestor of a Chachapoya genome, which is plainly ancient. But there’s nothing unusual about this at all, because it could of course be the case that the Mexican individual is the living representative of a people that predated and are the ancestors of the Chachapoyas. This is possible because mtDNA is so stable. Note that because the dataset has been diligenced to ensure provenance, if a genome is e.g., identified as Mexican, then the individual in question is ethnically Mexican, as opposed to simply located in Mexico (see Technical Notes below).


Technical Notes
I’ll begin by explaining in more detail how the algorithm actually works, and then close with some notes on the provenance of the dataset itself. As noted above, the core test is satisfaction of a simple inequality between three genomes. However, the algorithm has some optimization in it. Specifically, it begins by building clusters for each genome of the dataset, where a genome B is included in the cluster for genome A if genomes A and B have at least 99% of their bases in common. This builds a cluster of 99% matches for every genome in the dataset. It then fixes a genome C, and searches for the genome B, that minimizes the number of bases in common. Assume that genome B is found in the cluster for genome A. It then tests the inequality, which if satisfied, ensures that genomes A and B, and A and C, have more bases in common than genomes B and C. However, it also ensures that genomes A and B, and A and C, are a 99% match, and moreover, genome B minimizes the number of matching bases between B and C, over the entire dataset. As a consequence, this algorithm ensures that genomes A and B, and A and C, are a 99% match, and at the same time, ensures the maximum amount of mutation has occurred separating genomes B and C. Said otherwise, this ensures that genomes B and C are as dissimilar as possible, given the dataset, yet both mutually connected to another genome A, with which both have a 99% match. Finally, the code ensures that the result is either a tree, or a collection of trees (i.e., a forest), in that it precludes loops, and precludes parents from being linked to by children (note that the graphs are directed).
The dataset itself consists of 382 complete human mtDNA genomes from the NIH Database, taken from 33 ethnicities, including several ancient and archaic ethnicities. All of the genomes have been diligenced to ensure that the GenBank notes associated with the genomes imply the actual ethnicities, as opposed to just the location of the individual. That is, if the classifier for a genome is e.g., Chinese, then the GenBank notes explicitly state or plainly suggest that the sample is in fact from an ethnically Chinese person, as opposed to a person located in China.
The method of comparison involves counting matching bases, after a simple alignment that shifts the entire genome (if at all), to align it to what is plainly a globally common sequence of 15 bases. This is also apparently the default NIH alignment, which you can see for yourself by looking through their database. See e.g., these three genomes: Genome 1, Genome 2, and Genome 3, all of which contain exactly the same opening 15 characters. Because mtDNA is circular, this is plainly a deliberate starting point that we use as well, for simplicity. Finally, note that most of the genomes do not need to be shifted at all, because the NIH presents nearly all of the genomes I’ve seen using exactly the same alignment. Specifically, only 5 out of the 382 genomes in the dataset were shifted by an average of 2 bases.
The only thing I’ve learned studying genetics, is that nothing we think of is true in terms of race, and that all of our mothers, are probably severely disappointed in us.
Here’s the code:
https://www.dropbox.com/s/pmi56hjjugzcwrk/Generate_Heridity_Tree.m?dl=0
https://www.dropbox.com/s/1ikhuz6xnaesejh/Genetic_Furthest_Neighbor_Single_Row.m?dl=0
https://www.dropbox.com/s/y19d8ein5wjxe3a/Genetic_Alignment.m?dl=0
https://www.dropbox.com/s/nrczoxeqezvnls1/Genetic_Preprocessing.m?dl=0
https://www.dropbox.com/s/aeko80flttnk8b3/Genetic_ShiftbyK.m?dl=0
Here’s the dataset:












 bases).
bases).