I’ve always had two Ancient Egyptian genomes in my dataset, though I was only interested in the older of the two, which is from about 4,000 years ago, primarily because this is prior to Rome, and also because of the obvious morphological differences between Egyptian Royalty before and after Rome. Specifically, prior to Rome, the Egyptians looked Asian, and are in fact genetically related to Asian people, whereas afterwards, they looked European. On the left is Menkaure and Queen Khamerernebty II (c. 2,530 BCE), courtesy of MFA Boston, in the center is Nefertiti (c. 1,370 BCE), courtesy of Wikipedia, and on the right is Cleopatra (c. 50 BC), courtesy of Wikipedia, who plainly looks nothing like the rest of them. Moreover, the aesthetic is also completely different, plainly closer to that of Rome and Greece, and I would wager that the skills that led to earlier Egyptian art were lost, together with the bloodline, which explains the change in the people themselves, and their art.
I just tested the newer of the two genomes, which is from around 120 AD, and there are significant differences. Below is a plot of the differences between the two populations, with the newer Egyptian genome distribution subtracted from the older Egyptian genome distribution, and so a positive number in a given column implies the population in question is closer to the older Egyptian genome than the newer Egyptian genome, and vice versa. The minimum match count is set to 70% of the total genome. There were 105 other genomes that matched to the older Egyptian genome, and 87 other genomes that matched to the newer Egyptian genome. There is apparently no overlap between the two genomes, and together, 192 people are related to one or the other. There are a total of 484 genomes in the dataset, and so rough justice says about 40% of the world’s current population is related to the Ancient Egyptians, though this is not precise, because there are a significant number of other ancient genomes in the dataset. The net point is, especially because there are only two genomes, a large number of people alive today are related to the Ancient Egyptians, all over the world, and you can see that below.
Because these are single genomes, and because mtDNA is so stable, even over thousands of years, you can’t be certain that the differences between the two genomes are the result of time. Specifically, it could just be that these are two different families that are both from Ancient Egypt, and that both bloodlines were extant throughout Ancient Egypt. If this is the case, then it’s just chance that caused them to appear in this particular order. Note that I’m not referring to the order of their actual discovery (i.e., the date someone dug them up), and instead, the point in time at which the actual people lived. That said, the older of the two genomes is plainly significantly closer to ancient civilizations, specifically the Phoenicians and the Saqqaq. They are also significantly closer to the Thai people, which is consistent with the obvious fact that the Ancient Egyptians were visibly Asian people, prior to Rome. The newer genome is instead closer to the Swedes, Igbo (of Nigeria), the Icelandic, and the Munda (of India), who are all closely related to each other. This is again consistent with a migration-back to Africa hypothesis, which I’m now convinced of. If you’re interested in a more detailed analysis of these issues, and the underlying software, see my paper, A New Model of Computational Genomics.
All that said, the net takeaway is that a large portion of the world’s current population seems to have been a part of a very large group of people that lived literally all over the world, in antiquity. This suggests unambiguously, that ethnically diverse, and almost certainly interracial people, have existed for thousands of years, possibly longer, and the Pre-Roman Egyptians are themselves obvious evidence of this. Yet again, given the fact most modern people are far too stupid and lazy to build pyramids, anatomically correct yet innovative sculptures, and navigate the world in a small boat, the obvious conclusion is that these people were just better than you, but that’s not so hard, is it?
I’ve put together some code that allows us to compare entire populations, and find their Nearest Neighbors. The theory underlying the results is that mtDNA can be used to predict ethnicity with about 80% accuracy. See, A New Model of Computational Genomics [1], generally. As a consequence, it must be the case that mtDNA carries information about the paternal line of an individual, otherwise it would be impossible to produce such high accuracies. Because mtDNA comparison implies surprising genetic relationships among superficially disparate people, there must be an explanation for it. As it turns out, the methods presented in [1], are perfectly consistent with a migration back to the West from Asia, which finds support in academic literature [2], and moreover, I’ve presented perfectly sensible, and independent mechanical theories that explain this as well. So e.g., how could it be that a Norwegian and a Nigerian share a significant portion of both maternal and paternal lineages? The answer is that both likely migrated back from Asia, about 70,000 years ago (see [2] generally), and moreover, that appearance is coded for by a very small portion of the total human genome (at least that’s my hypothesis).
We can therefore, given two distribution of matches between two given populations, and many others, compare the two distributions For example, the chart below (on the left) shows the distribution of mtDNA genomes that are a 99% match to the Norwegian people. The y-axis shows the normalized maximum possible number of matches, and the x-axis shows the population name. The chart on the right shows the same for the Nigerian people. As you can plainly see, some Norwegians and Nigerians are a 99% match to the other, at least on the maternal line, as measured by mtDNA. And as noted, it must be the case that mtDNA carries information about paternal lineage as well, and therefore, we can also conclude, that these two individuals share a significant portion of their paternal lineages. There is simply no way around this, it must be the case, and assumptions to the contrary are the result of a culture that is limited in scope, simply because written human history is only a few thousand years old, whereas our actual history is at least several hundred thousand years old, and possibly millions of years old.
That said, the distributions are plainly different, and that is the purpose of this analysis, which is to take an entire population, and find the population that is most similar to it, as measured by their distributions. These results are less surprising, though some are still counter-intuitive. The table below shows the results of this analysis, which you can run for yourself using the attached code. The comparison between two populations is straightforward, you simply take the absolute value of the difference between two corresponding columns in the charts above. So for example, if we’re comparing Norwegians and Nigerians, we would take the height of column 1 in the left chart, and subtract it from the height of column 1 in the right chart, and take the absolute value of that difference. We then take the sum over all of the columns (i.e., their differences), and divide by 50, which is the maximum possible difference, since there are 50 populations in the dataset. This produces a measure of distance between the populations over [0,1], and below, you’ll find the Nearest Neighbor of each population (i.e., the other population with the minimum distance to the population in question). The results are sorted in order of increasing distance, and so the closest relationships are listed first. Note I’ve excluded any single-genome populations (e.g., the Ancient Egyptians), as well as the Denisovans, since the Denisovans are not a 99% match to any modern population, though they are a 70% match to many living populations.
You’ll see the Iberian Roma and the Papuans are the closest match. This might seem surprising, given that they’re plainly morphologically distinct people. However, they are both nearly perfect matches for Heidelbergensis, and as a general matter, living populations that are related to Heidelbergensis, are all close matches to each other (i.e., 80% or more of the mtDNA genome is identical). You’ll also see some obvious results, like the Scotts and the Norwegians, the Turks and the Greeks (despite the animosity), and the Khoisan and the Nigerians. However, you’ll also see some surprising relationships, in particular, the connection between the Khoisan and the English. In particular, both the English and the Khoisan are closely related to the Pre-Roman Egyptians, and the Saqqaq people of Greenland. Isn’t life something when you really do the work?
Here’s the table, the code is below, together with the dataset.
I’ve expanded my dataset of human mtDNA to include 14 Greek genomes, and 16 Turkish genomes. The results are interesting given the historical animosity, and violence, between Greeks and Turks, in that they are very similar people. In fact, the main reason I’m writing this note is because the Turks are closer to the Greeks genetically, then the Greeks are to themselves. That is, the match score between Greeks and other Greeks is lower than the match score between Greeks and Turks. Intuitively this sounds suspicious, but it actually makes perfect sense –
The Greeks are closer to the broader set of Europeans (and others) than they are to themselves, implying a heterogenous population of global people, including Turks. That is, the Greek population plainly includes a significant number of people that are basically identical to people that live in Turkey, and are ethnically Turkish, though they’ve likely forgotten this, and instead identify as Greek. Recall, all the genomes in my dataset are diligenced to ensure that the person in question is e.g., ethnically Chinese, as opposed to simply located in China. So in this case, all of the Turkish genomes are from people living in Turkey, that are identified as ethnically Turkish, and the same is true of the Greeks. Below you can find the distribution of 99% matches between the Greeks and everyone else (left), and the Turks and everyone else (right). As you can see, they’re similar populations. The population acronyms can be found at the end of my paper, A New Model of Computational Genomics (e.g., TK is Turkish).
In order to refine the analysis, I produced a third chart (below) that shows the difference between the match scores for each population, to get a better sense of the difference between Greeks and Turks. All I did was literally subtract the Greek match score for a given ethnicity from the Turk score for that same ethnicity, causing the range (i.e., y-axis) to vary from -1 to 1, though as you can see the differences are extremely small, and are instead bounded by -0.1 and 0.1 (i.e., 10%). This tells us that the distribution of global blood lines in Greece and Turkey are very similar (i.e., within 10% of each other), and you can see my earlier point, that the Turks are more Greek than the Greeks, in that the match score between the Turks and the Greeks is higher than the match score between the Greeks and the Greeks. And again, this makes perfect sense, if a large number of people with Turkish ancestry live in Greece, and nonetheless identify as ethnically Greek, due likely to forgotten history, which is obviously possible.
That said, you can see there are differences, just going in order, in that the Turks are closer to the Italians and the English. Moreover, the Greeks are significantly closer to the Basque (a group closely related to Northern Europeans), and the Turks are significantly closer to the Georgians. Further, the Greeks are significantly closer to Icelandic people (and all the other Northern Europeans) than the Turks, and this is really strange when you consider the fact that the Turks were part of the Axis Powers, despite the fact that they’re plainly not as close to Northern Europeans as the Greeks. Yet somehow, the Greeks were occupied by the ostensibly racially pure Axis Powers, in particular the Italians, who are obviously closer to the Turks genetically. It seems like WWII does make sense, it’s just the complete opposite of the public narrative sold by the Axis Powers. As a general matter, Nazi ethnic ideology is obviously complete nonsense, and the Europeans are unquestionably heterogenous people, and the Northern Europeans in particular, are arguably the most heterogenous people on Earth, with a mix of African, Asian, and European ancestry.
Consistent with this revealing analysis, the Northern Europeans, in particular the Danes and Norwegians, were also sent to concentration camps, despite not being Jewish. And not to pump my own work, but I’ve already researched this history thoroughly, so you can read all about it in the Afterword to my book VeGa. Though the numbers interned might seem small (there are plenty of links to primary sources in VeGa), keep in mind these are extremely small countries, by population. This is also consistent with the persecution of the Basque people during WWII, who are closely related to the Northern Europeans. It sounds insane, superficially, but I think the purpose of WWII was the annihilation of a group of people that all appear to be related to Europeans, including of course the Jews. I think this continues to this day. Common sense asks, who the Hell would do that? I think the answer is the Catholic Church, an institution that ruled Europe with an iron fist for almost two-thousand years, only to have Martin Luther, Henry the Eighth, Napoleon, and ultimately the U.S. marginalize the Catholic Church to the point of near irrelevance. And the Church was obviously involved in the Nazi Party from day one, and moreover, is also obviously involved in extremely high-level organized crime. My hypothesis is that the Church was going broke, science was obviously taking over, leaving the obedience the Church sought after for uneducated, unproductive people, who don’t have the money to support their extravagant lifestyle. To put it bluntly, they never cared about religion, the Church was and is a political and economic institution, so when Catholicism stopped paying the bills, they turned to crime, and I’m not joking at all, read God’s Bankers.
Amazingly, I came up with a Nigerian character for VeGa, who’s best friends with a Swedish guy, and as it turns out, the world being what it is, the Igbo people of Nigeria are extremely closely related to some Swedes, and seem to have identical histories as well. The Igbo people were of course also subject to genocide, and might have been targeted by the Trans-Atlantic Slave Trade. This character was not intentionally created (i.e., I came up with the character long before my work in genetics), and it doesn’t mean that people should use science as an excuse to be scientifically racist, and only friends with people that have your genetic lineage, even though this would produce a remarkably diverse world. The point is instead that the world is so complex, that things that sound stupid and racist, are probably stupid and racist. That said, it’s pretty obvious someone has it out for a very specific group of people, that is superficially diverse, but all seem to come from Asia. Moreover, there’s some support for the claim that the Nazis were using rudimentary genetics to select people for extermination. This would allow them to discern among e.g., Jews and Gypsies, killing only some of them, and it is also consistent with the mysterious and awkward fact that Scandinavians, that were not Jewish, were sent to concentration camps in large numbers. Many Scandinavians are of Asian descent.
I found an Igbo genome on the NIH website, and I just never got around to writing about it, though it’s positively fascinating. The Igbo people are (to my knowledge) located primarily in Nigeria, and when I initially started my work in genetics, I noticed some astonishing finds in Nigerian people, with some basically perfect matches to Norwegians and Japanese people. At this point, I’ve made perfect sense of how these things are possible, and they’re not at all inconsistent with accepted theories, they have nothing to do with slavery, and in particular, there’s academic support for a very early migration back to Africa from Asia about 70,000 years ago. My hypothesis is that many Northern Europeans were part of that same original group of people in Asia.
The Igbo genome suggests exactly this, since it is a 98% match to an Icelandic genome, many Swedes, and some Norwegians, Finns, and Danes (see left chart above). Moreover, if you reduce the match threshold to 90% of the genome, you find the Igbo genome is a match to the Munda people of India (see right chart above). This is perfectly consistent with the hypothesis of a migration back to Africa from Asia, which is again, at least supported by some academic research. Specifically, in this case, it suggests that the Ibgo are from India. You can supplement this with a hypothesis that the same original group of people from India separated, with some of them going to Scandinavia, and others going to Africa. Keep in mind, Iceland is far more insular than the rest of Scandinavia, so much so that Icelandic is a distinct language, that is close to Old Norse, a language from the Viking Age. And though I can’t find more than one Icelandic genome, common sense suggests the Icelandic people are extremely homogenous, and this is supported by the fact that inadvertent incest is a serious issue in Iceland. The point being that this is probably not a one-off, and moreover it can’t be, because the Igbo are plainly related to the Northern Europeans generally.
Black Tree brings the power of parallel computing, together with data compression, producing runtimes that are simply incomparable to other Deep Learning techniques. For a high-level academic summary of the underlying algorithms, see Vectorized Deep Learning. Download a Free Version of Black Tree from www.blacktreeautoml.com.
The results below were generated using Black Tree’s “Supervised Delta Classification” algorithm. This algorithm is included in the Free Version of Black Tree, so you can download the datasets below and see for yourself, that there is simply no contest between Black Tree and other Deep Learning techniques. All runtimes were generated on a MacBook Air 1.3 GHz.
Dataset
Classification Accuracy
Total Runtime (Pro)
Total Runtime (Massive)
UCI Credit 25,500 Training Rows 4,500 Testing Rows
83.33%
1,467 seconds
223.7 seconds
UCI Ionosphere 298 Training Rows 53 Testing Rows
94.11%
0.755 seconds
0.201 seconds
UCI Iris 127 Training Rows 23 Testing Rows
95.65%
0.237 seconds
0.086 seconds
UCI Parksinsons 165 Training Rows 30 Testing Rows
90.90%
0.379 seconds
0.083 seconds
UCI Sonar 176 Training Rows 32 Testing Rows
95.65%
0.513 seconds
0.108 seconds
UCI Wine 151 Training Rows 27 Testing Rows
96.70%
0.310 seconds
0.082 seconds
Image Classification
Black Tree’s image compression algorithms allow Image Classification tasks, including medical imaging classification, to be accomplished in roughly the same amount of time as Data Classification tasks, again producing simply unparalleled runtimes. Black Tree Pro and Black Tree Massive use the exact same image processing and classification algorithms. The Free Version of Black Tree includes the exact same algorithms, with a hard limit of 2,500 images. Accuracies and runtimes for the MNIST Numerical and MNIST Fashion Datasets are 99.95% and 286.09 seconds (5,000 Training Rows and 5,000 Testing Rows), and 92.85% and 15.90 seconds (1,000 Training Rows and 1,000 Testing Rows), respectively.
Black Tree Runs in a GUI
The front-end for Black Tree runs in an easy-to-use interface, reducing Deep Learning to a task that can be accomplished by an admin or assistant, thereby allowing for radical reductions to costs and headcount associated with Deep Learning. For the same reasons, Black Tree allows firms and individuals to spend a small sum of money (see pricing below) to test the question of whether investing in Deep Learning is worthwhile. For some users, this question can likely be answered by the Free Version of Black Tree.
Pricing
Download the Free Version of Black Tree, which includes (i) data classification and clustering, and (ii) image classification (grayscale only), up to exactly 2,500 rows / images (non-commercial license), or select from the commercial licenses below.
NOTICE: All sales are final, no refunds available. For technical support, see Contact Information.
A lifetime commercial license for one user, which includes (i) data classification and clustering, and (ii) image classification and clustering (grayscale only), up to around 25,000 rows.
A lifetime commercial license for one user, which includes (i) all of the algorithms included in Black Tree Pro, (ii) a significantly faster Supervised Delta Classification algorithm, (iii) a significantly faster normalization algorithm, together with (iv) Massive Algorithms that can classify 500,000 rows in approximately ten minutes, and (v) confidence metrics that allow for precise classification.
Black Tree Osmium (Coming Soon)
A lifetime commercial license for one user, which includes (i) preprocessing, compression, analysis, and anomaly detection algorithms that can be applied to data, image, video, and 3D and higher dimensional data and video (i.e., high-dimensional time-series), (ii) image and video classification algorithms, (iii) 3D object detection, tracking, and classification algorithms, (iv) high-dimensional time-series prediction and interpolation, (v) algorithms that can detect periodicity and stable average values in time-series data, and (vi) N-dimensional optimization.
I noted in my paper, A New Model of Computational Genomics [1], that many living human beings are very closely related to both Denisovans and Heidelbergensis. This likely doesn’t come across in standard genome comparison, because it seems current standard methods make use of local alignments, that effectively ignore insertions and deletions. This causes basically all living people to seem roughly similarly related to archaic humans. If you instead don’t use local alignment, you see drastic differences in match counts, with only some modern humans closely related to Heidelbergensis and Denisovans. First off, insertions and deletions are obviously very significant, since they produce diseases like Downs Syndrome and Williams Syndrome. Secondly, my methods are plainly more precise than using haplogroups (see [1] generally), and I use a single global alignment method. The net conclusion being, that local alignment produces imprecision because it ignores insertions and deletions, which are obviously significant. For example, run a BLAST search on this genome, and you’ll see “gaps” in the reports, which plainly artificially inflate the match percentage. In contrast, my work is able to predict ethnicity with roughly 80% accuracy, even between e.g., Swedes and Norwegians. This is simply not possible using local alignments, because everyone will end up a nearly perfect match to everyone else, which is obviously not true.
I’ve since updated the human mtDNA dataset that is the subject of [1], to include another 7 Denisovan genomes, and a population of 11 modern Kenyan genomes, as well as others. The dataset now contains a total of 452 complete human mtDNA genomes, over 47 global populations, including many ancient, and archaic humans. All genomes come from the NIH, and the dataset includes a provenance file with links to the NIH Database for each genome. All genomes have been diligenced to ensure that the person in question is e.g., ethnically Chinese, as opposed to someone simply located in China. See Section 1.4 of [1].
When running the same analysis I’ve always run, comparing the Denisovan population to the entire dataset, I found many Kenyans were a match. See the chart below, and see the end of [1] for a table with acronym names (e.g., KN is Kenyan). This is quite significant, since Southern Denisovan fossils have yet to be found, and if all hominins come from Africa, then there should be evidence of Denisovans in Africa. Unlike some of the surprising genetic relationships I’ve found, this could unfortunately be explained by slavery, for the simple reason that many Europeans are also matches to Denisovans. If Europeans related to Denisovans enslaved Kenyans, they could have reproduced with those Kenyans, which would have altered the maternal lineage of Kenyan people generally.
However, my work also allows us to determine whether a given genome A is the ancestor of another pair of genomes B and C. If modern day Kenyans are the genetic ancestors of the ancient Denisovan genomes collected in Asia, then this would eliminate slavery as a possibility, at least with respect to those ancestor genomes, and instead provide evidence for the claim that the Denisovans originated in Africa, and that at least some modern day Africans are the ancestors of the Denisovan fossils found in Asia. This is apparently the case, and moreover, the same is true of other Kenyans, with respect to Heidelbergensis. That is, some modern day Kenyans appear to be the ancestors of Denisovans, and others the ancestors of Heidelbergensis. The net conclusion is that all hominins could have come from Africa, and you can read this article for an explanation as to how this could happen, mechanically. This is obviously consistent with the popular narrative, but these things are not obvious, and there is both doubt and multiplicity in the academic community, though no one seems to think we come from America, so that point seems settled.
For an intuition as to how the ancestry test works, posit 3 genomes A, B, and C. Now assume B and C have more in common than A and B. A cannot be the ancestor of B and C, since that would imply that B and C both started out basically identical to A (since A is the ancestor of both B and C), and then both B and C randomly mutated to have more in common with each other than they do with A. This is basically impossible as a matter of probability, since it’s analogous to two people flipping independent coins ten thousand times, and ending up with nearly the same sequence of heads and tails. This is obviously wrong, but for a fulsome discussion, see Section 6.1 of [1].
The attached code applies exactly this test to two populations, treating one as ancestor (A), one as a descendent (B), and then attempts to find a genome C for which AB > BC and AC > BC, where AB, BC, and AC, are the match counts between genomes A, B, and C, respectively. If this inequality is satisfied, then it is at least possible for genome A to be the ancestor of genomes B and C. As noted, 2 Kenyan genomes out of 11 satisfy this test for the ancient Denisovan genomes, all of which were collected from ancient sites in Asia. Further, this inequality is also satisfied for 3 Kenyan genomes for the single Heidelbergensis genome included in the dataset. This seems to be unique to Kenyans, suggesting some Kenyans are truly ancient people. Specifically, 0 out of the 9 Nigerian genomes satisfied either test, and only 1 Khoisan genome out of the 10 Khoisan genomes satisfied the test for Denisovan, and only 1 Khoisan genome (a different genome) satisfied the test for Heidelbergensis. Note that the Khoisan are believed to be ancient people themselves. Further, 0 Egyptian genomes satisfied the test for Denisovan, however 5 Egyptian genomes satisfied the test for Heidelbergensis. The net point is again, all hominins seem to come from Africa, and a significant percentage of Kenyans (about 45%) seem to be truly ancient people.
I’ve also attached some extra code, that allows you to find descendent populations from a given population. You’ll note that all of you have committed atrocities against your own ancestors, I’m guessing because intelligence is not yet too common in the overall human population.
Posit a language over a set of characters capable of expressing any mathematical proof. Now consider the set of all strings that can be generated by taking any characters from . For example, , where each would be such a string in . The cardinality of is given by . Now consider the portion of comprised of strings that express a true theorem of mathematics. It is of course going to be extremely small, compared to the total cardinality of , which grows exponentially as a function of .
In my paper, Information, Knowledge, and Uncertainty [1], I showed both as a matter of theory, and empiricism, that the knowledge conveyed by an observation is given by,
,
where is the information content of the observation, and is the uncertainty of that observation. The information content of an observation is simply the logarithm of the number of states the observation could have taken on. See [1] generally.
Posit a theorem , and further assume that its shortest proof is of length , and that it is unique (i.e. there is no other proof of length ). It follows that the information content is in this case . The uncertainty is given by , because the state space of the problem is reduced to one possibility (i.e., the proof and theorem sought after). See Section 2 of [1]. It follows that the knowledge conveyed by is . Therefore, the knowledge conveyed by a theorem is a linear function of the length of its shortest proof.
Now you could of course argue that the calculation of is unduly generous, because mathematicians don’t churn through all possible strings. As a consequence, perhaps a more fair measure would be the number of valid statements of length . However, this is intuitively still exponential as a function of , though it is an interesting combinatorial problem. Nonetheless, the point being, that a standard page contains roughly 2,000 characters, implying an enormous amount of information is conveyed by theorems. This is contrast to the amount of information conveyed through observation, which is discussed at length in [1], and is plainly not as useful, because in order to produce an amount of knowledge equivalent to a 1 page proof, you would have to produce roughly 2,000 observations. No human being can consciously store 2,000 observations in their memory. In contrast, you can study, understand, and memorize a proof. This implies that mathematical knowledge allows human beings to maximize their measurable knowledge, whereas observation plainly has a low threshold, since you simply can’t keep too much information in memory.
The plain takeaway is that mathematicians produce knowledge that is plainly more useful than the information produced by ordinary people, and this is obviously the case anyway.
I have a ton of unpublished work on NLP, for the simple reason that I found absolutely no opportunities to make money from it, despite the fact I think it’s correct, though untested. However, it just dawned on me, that for the as of yet unfinished version of Black Tree, Osmium, which will include basically my full A.I. library, a GUI will probably be unmanageable, for the simple reason that my library is enormous – you can’t have a button for everything. As a consequence, it would probably be more efficient to simply type what you want Black Tree to do, using English. This is non-trivial, but I’ve already done enough work on NLP to make it happen. As such, I thought it worthwhile to at least introduce the basic concepts.
Specifically, every sentence has a subject, verb, and possibly an object. The subject, verb, and object, could all be qualified by other words, specifically, adjectives, adverbs, and quantities (e.g., some, all, one, the, etc.). This sounds trivial and obvious, but now you have an obvious algorithm for parsing a sentence – i.e., look for the verb, then look for the subject, and then look for the object (if it exists). Then look for their respective qualifiers (if they exist). This will cause every sentence (ignoring multiple independent clauses for now) to be reduced to a structure that contains three things, each of which could be associated with qualifiers. You can then compare a given sentence to a dataset of sentences, all stored in that format, which will give you meaning, if you simply return the set of sufficiently similar sentences. You can also give mechanical meaning to a sentence, which is my goal, by comparing a given sentence to a dataset of sentences that are associated with code.
First you load the sentence into a matrix, where each row contains a word, and compare every row to your dictionary, which contains articles of speech, thereby finding the verb. As a consequence, finding the verb in a sentence can be done in constant time, in parallel. Then once you find the verb, you search for its associated object, which must be a noun. You’re looking for all the nouns in the sentence, which again requires looking up each word in the sentence in a dictionary, to obtain its categorization as an article of speech. If you have an existing NLP dataset, you should be able to produce a best answer among all nouns in the sentence, for a given verb. For example, if the sentence is, “The drunk man ran to the dingy bar.”, then “man” is almost certainly going to be more frequently associated as a subject with “ran” as a verb, than “bar” and “drunk” (which can be a noun).
You can already see that it should be easy to produce a score for every noun in a dataset for every verb, which will allow you to quickly produce a best answer, if you calculate the scores beforehand. If the score’s already calculated beforehand, and stored (this will obviously be a big dataset), then it’s a constant runtime operation. In the worst-case, you have to calculate the score for all noun-verb combinations in a new sentence, for a given verb. This can be done by finding all (or at least some) instances of that noun-verb combination in the dataset, and calculating the percentage where the noun is the subject for the relevant verb (as opposed to the object). If the dataset is stored as a collection of matrixes (i.e., each sentence in the dataset is a matrix, with one word per row), then this can be done in two steps for each noun, in parallel (i.e., find the sentences that contain both the noun and verb in question). Then you apply the exact same process, except this time looking for an object, which will obviously return “bar”, rather than “drunk”. If you have independent clauses, then you find all the verbs separately, and apply an analogous process that will still work, because you’re finding the best pairs of subject / object and verb, you’re just doing it over a longer sentence that contains multiple verbs. In all cases, you have a constant runtime search for parsing a single sentence for meaning, and the total runtime will be a multiple of that constant runtime, given by the number of subject-verb, object-verb combinations, which should be low for any reasonable sentence, especially if it’s in a business context (i.e., an instruction or query).
Ultimately, what I’m planning to do, is have ML code be generated by NLP instructions, using already written ML snippets that are modified by the user’s instructions. This is literally what the GUI for Black Tree does, which is generate tailored ML code, using template code, as modified by the user’s selections. I would in this case be substituting the GUI with a set of instructions taken from an English sentence. This is not trivial, but it’s not that hard, at least using this methodology.
When I originally started my work in DNA, I was astonished to find that superficially distinct people (e.g., Nigerians and Norwegians), were 99% matches on their maternal line, as measured by mtDNA. See, A New Model of Computational Genomics [1], generally. That is, 99% of their bases are exactly the same. I mulled through the intuitive suspicions like slavery, but that line of reasoning started to fade quickly, as populations all over the world were again 99% matches, with absolutely no known history to explain it. For example, the people of Thailand and Japan (where there’s no significant history of slavery), are 99% matches with some people in Scandinavia and Africa. Moreover, many people in Scandinavia are 99% matches to a 4,000 year old Ancient Egyptian genome. Slavery simply cannot explain these outcomes, and I am not aware of any history that does. The conclusion I came to is that the world was global a very long time ago, due simply to sailing –
This makes perfect sense, and of course the history could be lost if it’s sufficiently ancient, which it seems to be. Consistent with this hypothesis, there is at least some academic support for a very early migration out of Africa, to Asia, and back to Africa [2], around 70,000 years ago, which could on its own explain these results, without sailing until much later (e.g., allowing for the eventual peopling of Japan and other Pacific islands, which plainly requires sophisticated sailing).
However, I also recently realized that it must be the case that mtDNA carries information about paternal lineage. This follows from the fact that mtDNA alone can be used to predict ethnicity with roughly 80% accuracy, over a dataset of 36 global ethnicities. See Section 5 of [1]. Chance implies an accuracy of . This accuracy is simply too high, unless mtDNA carries information about paternal lineage as well. This is distinct from being able to determine who a given person’s father is, and is instead, information about the paternal line of the person in general, which in turn allows you to predict ethnicity.
We are then confronted with the problem of morphologically distinct people, with the same maternal and paternal lineages. That is, two populations that have very similar distributions of maternal lines, probably have similar distributions of paternal lines as well. My work shows unambiguously that there’s a set of global populations that are 99% matches on the maternal line, which in turn implies that they are probably highly similar on the paternal line as well. However, these populations include Africans, Europeans, and Asians, who are obviously morphologically distinct people. How could this be if they are so genetically similar?
Complexity, Selection, and Competition
I think the answer comes from complexity theory. Specifically, coding for color (e.g., skin or eyes), texture (e.g., hair), and quantity (e.g., size or height), requires very little information compared to coding for structure. Human beings are all structurally the same, and as a consequence, there shouldn’t be much variation in the genetics that codes for overall morphology. Similarly, because color, texture, and quantity are low-information variables, the portion of the genome that codes for these properties will be small relative to the size of the human genome as a whole. In contrast, the brain, nervous system, and sensory organs (e.g., the eye) are incredibly complex systems. As a consequence, they must require significantly more bases to code for than e.g., skin color. Said in simple terms, you can encode variables like size using integers, whereas coding for structure requires information about position and function, which is far more complex, especially for a system as complex as the brain. Keep in mind, the human brain is, as far as we know, the most complex system in the Universe.
You can then ask, why would Nature use efficient codes? And the answer there is that Nature is the most ruthless enforcer of efficiency, and probably why human beings even considered efficient coding in the first instance (i.e., it is the result of competition). Specifically, posit two otherwise identical species A and B. Species A uses a small portion of its genome to code for simple systems in the body, whereas species B uses a large portion of its genome to code for simple systems in the body. The larger the portion of the genome that codes for a given system, the greater the multiplicity of outcomes (i.e., the greater the number of variations that are possible for that system).
This is the case because there are a greater number of sequences that follow from a longer sequence of bases. Note that the number of possible sequences of length is , and as a consequence, the number of possible sequences grows exponentially as a function sequence length. It follows that species A will reserve a larger portion of its genome for more complex systems, thereby allowing for exponentially greater multiplicity in complex systems. This in turn implies that species A will be more diverse with respect to complex systems like, e.g., the brain, than species B. That is, you have an exponentially larger number of possible brains in species A than you do in species B. For the same reason, species B will allow for greater multiplicity in less complex systems, but this is obviously a waste. Therefore, species A will create more opportunities for selection on the basis of complex systems like the brain, and in the long run, species A will plainly outperform species B.
Genetic Similarity, Morphological Distinction
The logical conclusion is that the reason you have high genetic similarity among morphologically distinct populations is that they have similar brains, sensory organs, nervous systems, and complex systems generally, which should produce similar preferences. The portion of the genome that causes them to appear physically different is likely minuscule when compared to the total genome size. This could explain how e.g., an African person and Asian person, or Northern European person, could all be 99% matches, and as a general matter, share a significant portion of their total genome. More generally, the visible portion of the human body is plainly not the most complex part, nor is it the bulk by mass – the inside is. This implies a question for empirical testing, that is now possible to answer, specifically, whether whole-genomes follow mtDNA. If this is the case, which I suspect it is, given the fact that mtDNA alone can reliably predict ethnicity, at the level of a modern sovereign boundary, then the story of humanity needs to be rewritten, which will in turn change our understanding of not only the present, but history as well. That is, it’s impossible that these genetic connections arose spontaneously, and so there must be a history that brought them to fruition, which implies a very early, and very diverse world.
Historical Implications
As a consequence, our understanding of history is almost certainly wrong, based upon genetics, and also just common sense observation. One glaring example is the Ancient Egyptians, who were visibly Asian people, that seem to have straight hair and somewhat almond eyes, and I suspect based upon genetics and common sense, that they come from Nepal, since they are a 99% match to many modern Nepalese people. And there are other modern day Africans that look very similar, e.g., the Khoisan people, who are also in many cases part of the same global group of people that are mutual 99% matches to each other. If I had to wager, I’d say that we don’t have a good understanding of very early Ancient Egyptian history (i.e., beginning around 10,000 BC), and that the Egyptians initially came from Nepal, and might have been seafaring people for a very long time, prior to forming what we now know as Ancient Egypt. Again, this is consistent with [2], that argues for a migration back to Africa from Asia, around 70,000 years ago.
Menkaure and Queen Khamerernebty II, courtesy of MFA Boston.
Whole-Genome Sequencing
Some of this plainly doesn’t come across in traditional genetic research, which focuses on genes, and other signatures in genomes that are statistically common in populations. However, I did reach many of the same conclusions as researchers using traditional techniques (e.g., a migration back to Africa from Asia). Therefore, at the risk of being immodest, because my results are consistent with, but more precise than, traditional genetics, I think it’s fair to conclude the methods introduced in [1] are in general superior. Now, that said, being able to sequence entire genomes is relatively new, and so there is a practical explanation for this, which is that you have limited time and resources, and so you focus on a portion of what is in all honesty a gigantic mathematical object (i.e., the entire human genome). However, my work allows for whole-genome comparison and analysis in polynomial time, and was conceived of after the advent of whole-genome sequencing. As a result, we can now compare entire genomes, even on consumer devices, and therefore, ask questions about whole-genomes.
Image courtesy of Wikipedia
Moreover, there’s simply no way that traditional genetic research using Haplogroups will produce the kinds of accuracies my work produces. You can see this in the map above, which shows the global distribution of different Haplogroups, which plainly span large geographic areas. In contrast, my software is able to, e.g., distinguish between Norwegians, Swedes, and Finns, again with 80% accuracy, given a dataset that includes 36 global ethnicities. You can see that it is impossible to do this using Haplogroups alone, because it’s sloppy, and breaches national boundaries. If you want to understand exactly why my methods work better, read [1], but for an intuition, you’re starting with a gigantic mathematical object, a genome expressed as a vector of labels, that is potentially millions of characters long. Then, you’re searching for individual, presumably sequential signals that are common to a population. First off, the better signals might not be sequential, and my research suggests instead the best signals are randomly spread over a genome. See Section 7 of [1]. Secondly, even if the best signals are sequential (which is probably not true), there are an enormous and certainly intractable number of sequences to consider, because you have to subdivide populations, because every population is heterogenous (i.e., there are multiple bloodlines in every population). Therefore, you are basically guaranteed to miss some signals that are common to a population, producing the imprecise results above.
Application to Data
Attached is code that allows you to A/B test populations, and identify where on the genome their bases differ. It also outputs the average number of matching bases. As an example of the theories above, I compared a single Mongolian genome to a single Thai genome, and the number of matching bases is 5,028. Keep in mind that chance implies a match count of one quarter of the genome, which is 4,144 bases. As such, the Mongolian genome and Thai genome have little more than chance in common. I then compared 4 Thai genomes, to the full dataset of ethnicities, and the results are plotted below. The x-axis shows the population acronym (e.g., MN is Mongolian). The full table of acronyms can be found at the end of [1]. If a given Thai genome is a 99% match to e.g., a Norwegian genome, a counter is incremented. The y-axis shows the value of that counter for a given population as a percentage of its maximum. For example, there are 4 Thai genomes, and 20 Norwegian genomes in the attached dataset. As a consequence, the counter for the Norwegian population has a maximum of 80 (i.e., 20 x 4). The chart below shows the value of this percentage for each population on the y-axis, and in the case of the Norwegian population, it is exactly 13.75%. As you can see, there’s a very weak connection between the Thai genomes and the Mongolian genomes, of which there are 19, with a percentage of 2.63%. The plain implication is that despite superficial similarities, Thai people are much closer to Norwegian people, than they are to Mongolians.
In addition, there is a single Saqqaq genome in the dataset, and so 50% of the Thai genomes are a 99% match to that single Saqqaq genome. The Saqqaq were indigenous people that lived in Greenland from around 2,500 BCE to 800 BCE. Greenland is plainly geographically remote from Thailand, and moreover, requires a boat to get to – you simply cannot credibly claim that people can swim through the frozen waters around Greenland, in appreciable numbers. As a consequence, it must be the case that at least some seafaring capabilities existed in indigenous peoples during antiquity, suggesting at least the possibility of sophisticated seafaring people elsewhere. Moreover, the Ancient Egyptians were obviously very sophisticated people, and so they’re a decent candidate for the peopling of the Pacific, which obviously required sophisticated boats, and probably telescopes.
The distribution of match count percentages for the Thai population
Again, as you can plainly see, many Norwegians are a nearly perfect match to the Thai people. In fact, the maximum match percentage between Norwegians and Thais, is 99.76% of the full genome. This could explain why there are plainly Asian-inspired structures in Norway (and some other parts of Europe) known as Stave Churches, that obviously resemble Thai temples. Note that this is also consistent with the hypothesis that genetically similar people should prefer the same aesthetics, since they have similar brains and sense organs. The conclusion being that despite dissimilar appearances, the Thai and Norwegians are very closely related, whereas the Mongolians and Thai are not closely related. This is contrary to what are plainly racist, unscientific categorizations of people, revealing instead real, and deep genetic connections between superficially dissimilar populations.
A Norwegian Stave ChurchA Thai Temple
The Origins of Diversity and Humanity
I recently noted that it’s basically impossible for random mutations to persist, unless they occur on a mass scale in a given population. I posited that contagious microorganisms are the cause, specifically that microorganisms spread in a given population and cause similar mutations to that population. If those mutations are beneficial, selection will cause them to persist. This is also consistent with a common origin of multiple hominin species out of Africa, since otherwise, there’s really no good explanation for multiple similar hominin species to emerge from the same place. Mathematically, having the same significant random mutation occur twice has a probability of roughly zero, since the probabilities are governed by the Binomial Distribution, which means we should have at most one species of hominin, and there are instead many. In fact, there should have been at most one original human, who would be therefore incapable of reproducing. This idea is obviously wrong. The idea that they should all come from the same place without cause is therefore totally absurd.
If you instead posit the existence of microorganisms somewhere in Africa that cause great apes to mutate into hominins, then you can easily explain the emergence of hominins. This would also explain why e.g., Mongolians, some of whom are closely related to Heidelbergensis, look very similar to other Asians, who are not related at all to Heidelbergensis (e.g., the Thai people). That is, the microorganisms in Asia cause the relevant mutations that change morphology. The work above implies that a very small portion of the genome is responsible for appearance, and it is therefore perfectly plausible that the same mutation occurs to totally distinct people, on a mass scale, causing them to develop a similar appearance, while otherwise having very little in common genetically. However, the portion of the genome separating humanity from the great apes is presumably significant, and as a consequence, it should not occur as often. That is, it should occur more often than chance, because it has a cause (i.e., microorganisms in Africa), but it should occur less often than the mutations that change appearance in Asia, because the portion of the genome involved in appearance is presumably much smaller than the portion involved in transitioning from great ape to hominin.
If less than all of the mutations required to transition from great ape to hominin are effected, then I would wager the organism in question ends up with a genetic disease, and dies off. Similarly, if less than all of the mutations required to transition into an Asian morphology are effected, I would again wager the individual in question ends up with a genetic disease, and dies off. Because there are fewer genes involved in transitioning to an Asian morphology than there are in transitioning from great ape to hominin, the probability of a fatal error should be lower, since the number of mutations is much smaller (i.e., each mutation carries some probability of error, and so the total error is a function of the number of mutations). As a consequence, Asians should look roughly the same, and they do, since it is a “safer” mutation than transitioning from great ape to hominin. Note that I am plainly not considering e.g., Indians in this discussion, and there are of course other populations in Asia that are morphologically distinct, but this is not inconsistent with this hypothesis. In fact, it supports the hypothesis, because it shouldn’t happen all the time, just more often than great apes that transition into hominins, and greater than chance, which is plainly the case, given that a simply enormous number of Asian people have very similar morphology. This, despite the fact that they are plainly not all closely related as a matter of overall genetics.
I was introduced to Williams Syndrome through my research on genetics, and it is rare among genetic diseases in the sense that it’s not all depressing, and in fact, people with Williams Syndrome are incredibly charming, emotive, and in some cases articulate people. The disease is the result of deletions on Chromosome 7, and Chromosome 7 is believed to be connected to language skills, and possibly socialization itself. Amazingly, people with Williams Syndrome are simply incredibly kind people, and some are brilliant musicians. As a general matter, people with Williams Syndrome have a great affinity for music, even if they are not themselves musicians, plainly suggesting a connection between musical aptitude and Chromosome 7. I didn’t look too far into the particulars, but I did come upon a video interview with a simply charming woman named Alexandra, who has Williams Syndrome. Williams Syndrome is at times associated with cognitive difficulties, but simply watching Alexandra, you can tell that she has basically none, and is instead, quite articulate, and moreover, she has a high level of self-awareness, and can describe her emotional state in detail, despite her disabilities. In particular, she said something that stuck with me, which is that she loves looking in the mirror, and I do as well, though not because I think I’m the most handsome man in the world, but because of an innate sense of not being alone as a consequence of simply looking in the mirror. And in fact, whenever I brainstorm, I always look in the mirror, as if I’m having a conversation.
Schizophrenics often have hallucinations that cause them to dislocate their consciousness, and assign it to a part of their body that makes no sense. So e.g., they might think their consciousness lives in a book across the room. This is obvious literally insane, and cannot possibly be physically true. However, I think it is the result of what are literally multiple functioning consciousnesses in one brain. This is not metaphysical, and instead, I think the brain of a person with Schizophrenia is literally subdivided, into two consciousnesses. This does not mean two brains, or two copies of the entire brain, but instead, multiple instances of the portion of the brain responsible for consciousness itself, not the rest of the brain. Because consciousness is physically real, there must be a cause for it, presumably in the brain, and as this mechanism develops, presumably during childhood, it could splinter into multiple instances, creating multiple consciousnesses in one brain.
This would allow for literal, self-awareness, in the sense that one region of the brain responsible for consciousness observes another region responsible for consciousness. This is in essence parallel computing, with communication between the UTMs, which can be accomplished simply through a single shared memory, which is obviously critical for any functioning human being. In fact, that could be one of the things that goes wrong with Schizophrenia, leading to proper multiple personalities, due to memory unique to one portion of the brain.
It would also allow for arbitrary scaling of consciousness, which in this view would scale the potential for literally parallel thoughts, and therefore faster more efficient thinking. This could explain how some people solve seemingly non-computable problems, through potentially arbitrarily large arrays of consciousness that are going to be difficult to describe in words, because by definition, you have multiple independent sequences of thoughts. If the mechanism of consciousness is in the brain, but driven and perhaps even housed in a field (e.g., the electrostatic fields in the brain generate a magnetic field literally separate from the body), then you could even have infinite independent consciousnesses in one brain. This sounds far out, but it’s not, because the shape of the field determines the number, and if the shape of the field is infinitely divisible, then you could have a shape with an infinite number of discrete components. Such a mind would be strictly superior to a UTM, which is obviously the case for some people.
Returning to Williams Syndrome, I think Alexandra experiences exactly that, i.e., another person in her mind when she looks in the mirror, albeit in a manner that is not destructive to her psyche, suggesting a connection between Williams Syndrome and Schizophrenia, and therefore Chromosome 7. This could explain why musicians (and Alexandra) really are unusually happy, energetic people:
They’re literally never alone.
And although it is only anecdotal, at the same time, it’s clear there’s a connection between creativity and madness, in that Quincy Jones’ mother was schizophrenic, and he is of course himself, plainly a brilliant musician, and Paul Erdös, Sir Isaac Newton, John Nash, John Nash’s son John Charles Nash (again implying heredity), Erik Satie, and Caravaggio all suffered at times from mental illness, possibly Schizophrenia. Caravaggio actually murdered a man, over a tennis game, indicating that he was plainly insane, despite the fact that he was a genius. This comes across from Alexandra, who cannot stand being alone, experiencing anxiety as a consequence. The net point being, there seems to be a connection between self-awareness, which comes across as emotional intelligence in the case of Alexandra, and Chromosome 7, and in turn, a connection between self-awareness and parallel computation, which could explain the nature of genius, and its lamentable connections to madness. Expressing this mechanically, the deletions on Chromosome 7 associated with Williams Syndrome, and musical and creative aptitude generally, and perhaps Schizophrenia, cause multiple instances of the mechanism responsible for consciousness in the brain to develop, causing the individual to literally think differently than normal people.
My list of crazy people is plainly anecdotal, but I thought it was worth noting that Erik Satie is the only musician on the list. This caused me to consider potential causes, under the assumption that musicians are less likely to be mentally ill than other geniuses. This is obviously counter to the popular perception of musicians as degenerates, but those people are popular musicians, and they are degenerates. In contrast, to knowledge, none of Mozart, Brahms, Bach, Liszt, Prokofiev, Chopin, Faure, Chausson, Debussy, and Ravel suffered from any mental illness at all, and in fact, they were all really well-adjusted, productive people. This led me to the primary difference between music and all other art forms, which is that music requires physical discipline over time, to play a note only at the exact right moment, in the exact right place, in the exact right manner. This plainly requires a regulatory function in the brain that prevents impulses from translating into errors. As a consequence, I think it makes perfect sense that musicians would be among the most well-adjusted geniuses, for the simple reason that they by definition have a high degree of physical discipline, which will prevent them from, e.g., stabbing a man over a tennis match. At the same time, because music is quantitative, I don’t think it’s any less challenging at the highest levels, than mathematics. And so, fine art musicians would be primary targets for genocide by people seeking to damage demographics, since they plainly posses high intellect, but are not degenerates.















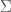 capable of expressing any mathematical proof. Now consider the set of all strings
capable of expressing any mathematical proof. Now consider the set of all strings 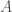 that can be generated by taking any
that can be generated by taking any 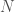 characters from
characters from  , where each
, where each 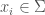 would be such a string in
would be such a string in  . Now consider the portion of
. Now consider the portion of 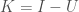 ,
,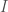 is the information content of the observation, and
is the information content of the observation, and 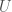 is the uncertainty of that observation. The information content of an observation is simply the logarithm of the number of states the observation could have taken on. See [1] generally.
is the uncertainty of that observation. The information content of an observation is simply the logarithm of the number of states the observation could have taken on. See [1] generally. , and further assume that its shortest proof is of length
, and further assume that its shortest proof is of length 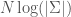 . The uncertainty is given by
. The uncertainty is given by 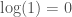 , because the state space of the problem is reduced to one possibility (i.e., the proof and theorem sought after). See Section 2 of [1]. It follows that the knowledge conveyed by
, because the state space of the problem is reduced to one possibility (i.e., the proof and theorem sought after). See Section 2 of [1]. It follows that the knowledge conveyed by 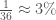 . This accuracy is simply too high, unless mtDNA carries information about paternal lineage as well. This is distinct from being able to determine who a given person’s father is, and is instead, information about the paternal line of the person in general, which in turn allows you to predict ethnicity.
. This accuracy is simply too high, unless mtDNA carries information about paternal lineage as well. This is distinct from being able to determine who a given person’s father is, and is instead, information about the paternal line of the person in general, which in turn allows you to predict ethnicity. , and as a consequence, the number of possible sequences grows exponentially as a function sequence length. It follows that species A will reserve a larger portion of its genome for more complex systems, thereby allowing for exponentially greater multiplicity in complex systems. This in turn implies that species A will be more diverse with respect to complex systems like, e.g., the brain, than species B. That is, you have an exponentially larger number of possible brains in species A than you do in species B. For the same reason, species B will allow for greater multiplicity in less complex systems, but this is obviously a waste. Therefore, species A will create more opportunities for selection on the basis of complex systems like the brain, and in the long run, species A will plainly outperform species B.
, and as a consequence, the number of possible sequences grows exponentially as a function sequence length. It follows that species A will reserve a larger portion of its genome for more complex systems, thereby allowing for exponentially greater multiplicity in complex systems. This in turn implies that species A will be more diverse with respect to complex systems like, e.g., the brain, than species B. That is, you have an exponentially larger number of possible brains in species A than you do in species B. For the same reason, species B will allow for greater multiplicity in less complex systems, but this is obviously a waste. Therefore, species A will create more opportunities for selection on the basis of complex systems like the brain, and in the long run, species A will plainly outperform species B.



