It sounds superficially impossible, but my work shows unambiguously that paternal selection impacts mtDNA, for the simple reason that males appear to select females on the basis of maximizing the number of bases they have in common with their female mates. Therefore, if you replace an existing male population with a new male population, the original female population will be selected for on the basis of maximizing the number of bases they have in common with the new male population, which will cause the lower-matching females to die off. See Section 5 of A New Model of Computational Genomics [1]. This will over time cause the two maternal lines to converge (i.e., the maternal line of the new paternal line will converge with the existing maternal line).
Today I developed a new technique that looks at the gaps in matches that occur as a result of insertions and deletions. That is, if you align to genomes, and then shift one of them by a single base at index i, the match count along indexes i + 1 through N (i.e., the genome size) will be reduced to chance. The question is, is there selection in those gaps? The answer is plainly yes, and by testing for it, you can determine paternal ancestry. Specifically, start with a genome, and then compare that genome to every genome in a given population (e.g., a single Ancient Egyptian genome compared to every Norwegian genome in a dataset). If Norwegians played a role in selecting Ancient Egyptian women (probably not true), then in the gaps, we should find evidence of selection, which would manifest as a number of matches that exceed chance. We can then measure the density of these above-chance matches within the gaps, which is consistent with selection by males from that population. Note that the criteria of materially exceeding chance provides an objective test, which is attractive, because you don’t have such a test in the non-gaps.
Doing exactly this for a Pre-Roman Ancient Egyptian genome, produces the distribution above, suggesting that the paternal line of the earliest Egyptians is from Asia, specifically, the ancestors of present day Javanese (JV) and Solomon Islands (SI) people. The full list of acronyms is included in [1]. The Ancient Egyptian maternal line is unquestionably from Asia, specifically the ancestors of modern day Thai people. See [1] generally. This is obvious when you look at the earliest Egyptians, who were visibly Asian, some of whom seem to have had straight hair. The code to produce the distribution above is attached below, and the dataset and any missing code is attached to [1]. Below are Menkaure and Khamerernebty II (circa 2532 BCE), courtesy of Boston MFA, both of whom are visibly Asian people. Overall, this algorithm provides even more support for the hypothesis that many modern Europeans and Africans are actually from Asia. Specifically, the Nigerians have Asian and Classical paternal lines (e.g., Sri Lankan and Phoenician), and the Classical people all also seem to be from Asia, not just the Egyptians. This is consistent with fairly recent academic results showing that there was an even earlier migration back to Africa, from Asia, about 70,000 years ago. My hypothesis is that this happened more than once, and that humanity began in Africa, then migrated to Eurasia and Asia, with many groups coming back to Africa, and some going to Europe, in particular Scandinavia.
I stumbled upon a simply incredible resource, specifically a database of ancient mtDNA maintained by Czech academics. All of the genomes have provenance files, with links to the source. Moreover, they all have dates, and locations, which is simply fantastic. I’ve been running analysis all morning, and one thing is clear: ancient Europeans are from Asia, specifically, they are related to the people of modern day Sri Lanka. You can run my genetics software on, e.g., this genome, which is from Belgium, around 33,000 BCE, and it is a 99% match to the Sri Lankans, and the Phoenicians (who are obviously related to the Sri Lankans). You find exactly this throughout history, in Germany, Austria, and Italy, suggesting that people came to Europe from Asia a very long time ago. Moreover, many Africans, specifically the Nigerians, are also closely related to the Sri Lankans, and Asians more generally. This is all consistent with a migration-back hypothesis, where humanity began in Africa, migrated to Asia, and then migrated back to both Europe and Africa.
Moreover, the second find is that the Roma people are definitely Russian, not Indian, which I already suspected based upon the fact that nearly a majority of Russians are a 99% match to the Iberian Roma. I’m not doubting that they spent a lot of time in India, and it seems reasonable that their culture developed there, however, their ultimate genetic origin is Siberia. Specifically, there’s a Siberian genome from about 45,000 BCE that is a 99.5% match to the Iberian Roma, and many modern Russians. Moreover, I’ve found no evidence of Roma people in Europe even up to the Middle Ages, except in Iron Age Finland and Ötzi the Ice Man. Both suggest that the Roma were originally Arctic people, and moreover, suggesting as a general matter, that the Roma people were fairly recent arrivals in Europe, at least in the numbers present today, which are significant (i.e., you have many people that are definitely of Roma descent that don’t declare it in genetic studies).
ResearchGate has for some reason deleted everyone’s project updates, which is really annoying, since a lot of work went into that in my case. Nonetheless, they thankfully allowed for the updates to be downloaded as html files that look basically the same. Here’s a link to all of my updates, which you can download as a zipfile.
A while ago, I wrote an algorithm that calculates the distribution of bases at each index of a genome, over a given population. For mtDNA, there are 16,579 bases in each genome (in the dataset attached below), and the algorithm calculates the density of Adenine (A), and Thymine (T), Guanine (G), and Cytosine (C), at each index, over an entire dataset. This associates each of the 16,579 bases, with 4 real numbers, each in , that sum to 1, for the simple reason that the bases must be one of the four. However, note there are minor deviations due to missing bases. The distribution of bases over the entire dataset are unequal, and that’s interesting in and of itself. However, I just tested the distribution of bases within each population ethnicity, to see if there’s meaningful variation in the distribution of bases. It turns out there is meaningful variation, suggesting at least the possibility that different populations select for different distributions of bases.
Specifically, the method I employed first calculates the distribution of bases at each index in a given population. It then calculates the overall distribution of bases within that population, producing four numbers, that represent the densities of the four bases, within that population, that should generally sum to 1, save for missing bases. This is done for each population, and there are 76 populations in this dataset, producing a 76 x 4 matrix. I then calculated the standard deviation in each column, as a measure of the variation in the densities of each base, across each of the 76 populations. The standard deviations of the distributions of A,T,G, and C, across all populations, are 0.0464%, 0.0367%, 0.0295%, and 0.0914%, respectively. These are small percentages, but they’re not uniform, and moreover, they’re not totally negligible quantities.
One initial observation, the Guanine-Cytosine bond is the strongest, and G is plainly more variable than the other bases. This is consistent with selection around the density of Guanine and Cytosine. Why would the distribution of bases matter? Well, the cytoplasm contains at any given moment a fixed distribution of bases, from which all genomes will draw upon to effectuate replication. As a consequence, there is by definition competition among genomes, and perhaps other parts of the cell, for the free-floating bases in the cytoplasm. As a consequence, the distribution of bases within a genome could impact cellular function and health. If the supply of bases in the cytoplasm is adequate to effectuate replication without any shortfalls, then an excess of a particular base in any genome, would cause an excess of that base in the cytoplasm. This could in turn affect cellular function and health. If the supply of bases in the cytoplasm is not adequate to effectuate replication without any shortfalls, in light of an excess of a particular base in a given genome, then this could again impact cellular function and health. As a general matter, intuition suggests a parity between the supply of bases in the cytoplasm and demand for bases based upon the distribution of bases in the genomes generally. However, as noted, even in the absence of such a parity, the distribution of bases in the genomes and the cytoplasm could impact cellular function and health.
This however does not explain why there we would be greater selection for Guanine-Cytosine bonds (i.e., the standard deviation is significantly higher for C), and moreover, why the standard deviation for C is the lowest of them all. One simple explanation is that there’s a handedness to the genome, and that the side being read by the sequencer is at least partially determinative of the distribution. Note that the opening sequence of bases in the dataset below is the same, and as a consequence, the sequencer is always reading the same side of the genome. One simple theory to explain the handedness of mtDNA is that it’s circular, and as a consequence, the genome has an “inside” and an “outside”. Now of course the other side of the genome must have a corresponding distribution (i.e., the other side of the genome must have a highly variable density of Guanine). However, the point is, the variability of Cytosine is much higher than the other bases, and as a consequence, it’s at least consistent with greater selection for Guanine-Cytosine bonds. The Guanine-Cytosine bonds happen to be the strongest (i.e., they are stronger than Adenine-Thymine bonds), and so it’s perfectly reasonable that they e.g., impact the ease of replication, and the integrity of the genome generally, which could present both advantages and disadvantages.
Note that the distribution at a given index in the genome will not directly determine the total distribution of bases in the genome. That is, if you e.g., swap two indexes in the genome, the overall distribution is unchanged, and therefore, all of the arguments presented above regarding the distribution in the cytoplasm are still true. However, this does not rule out selection at each index, since as noted, Guanine-Cytosine bonds are stronger than Adenine-Thymine bonds, and as a consequence, there could be advantages and disadvantages with respect to the integrity of the genome, based upon the distribution of bases along the genome indexes. This is a fascinating and to my knowledge, unexplored corner of genetics, that relates to selection based upon the overall distribution of bases (for purposes of cellular function), and their location along the genome (for purposes of genome integrity). This could explain the empirical fact that statistical imputation for mtDNA is categorically superior to sequential imputation. See Section 7 of A New Model of Computational Genomes. That is, there is significant selection taking place based upon whole-genome functions (i.e., random bases), beyond protein production (i.e., sequential bases). This suggests at least the possibility that whole-genome functions are more aggressively selected for than protein production. At first this sounds counter-intuitive, but it makes perfect sense:
If the cell can’t function, and the genome’s not stable, then it doesn’t matter what proteins are coded for, because that’s like writing on a burning piece of paper.
Putting it all together, once a population-specific mutation occurs, the genome’s integrity as a whole is subject to the possibility of deterioration, for the simple reason that a complex set of bonds has by definition been disrupted. This should therefore, cause intense selection in the rest of the genome, that allows for the mutation to exist in a structurally sound context. This would cause whole-genome selection, and explain the empirical fact that statistical imputation is stronger than sequential imputation, at least with respect to mtDNA. Moreover, the arguments above regarding the cytoplasm support the same hypothesis. If e.g., a mutation drastically changes the distribution of bases in a genome, then replication will draw differently on the bases floating in the cytoplasm, potentially causing disruptions to cellular function and health. As a consequence, whole-genome selection could remedy any drastic changes to the draw on the distribution of bases in the cytoplasm. However, unlike the structural integrity of a given genome, which requires whole-genome selection, the overall distribution of bases in the cytoplasm could itself adjust to the mutation, or other genomes could adjust to the mutation.
I’ve shown empirically that mtDNA is selected for, in particular by male humans when selecting female mates. See A New Model of Computational Genomics, in particular, Section 5. The data suggests quite plainly that males are selecting females on the basis of having overall similar mtDNA genomes. I didn’t explain how it is that males or females would know this, though I now think the answer is, that mtDNA is deeply connected to overall health. Therefore, it makes perfect sense for a species to develop a means of communicating information about mtDNA, since doing so, would allow for better mating decisions, and therefore, healthier offspring. I don’t know what the specific mechanism is, but I’d wager mtDNA actually does impact appearance in some subtle way that informs attraction between mates. This would in turn allow males to select females on the basis of having a similar overall mtDNA genome.
It’s not exactly a secret that both Nazis and Communists killed and persecuted intellectuals. The question is, did this have a meaningful impact on the demographics of countries occupied by Nazis and Communists? This answer is a clear yes, in my opinion. To understand why, the first component is that many modern humans are a 70% match on the maternal line with Denisovans, as measured using mtDNA. See my paper, A New Model of Computational Genomics [1], generally. This is not unique to Denisovans, and in fact, basically all Iberian Roma and all Papuans are a 96% match to Heidelbergensis, and many people globally are a 96% match to Neanderthals. See [1] generally. However, approximately a majority of people have no real relationship to any archaic humans, beyond chance (i.e., about 25% of the genome in common). All of this is based upon the dataset linked to in [1], which at this point consists of 600 complete human mtDNA genomes, all taken from the NIH Database. The most current dataset is linked to below, and an older version is linked to in [1].
The root observation is that many Jews (both Ashkenazi and Sephardic) are a 70% match to Denisovans, which you can see in the chart above. Note that all of the population acronyms (e.g., KN stands for Kenya) can be found at the end of [1]. Because so many Jews were killed during the Holocaust, and many Jews are technically Denisovan, we would expect countries occupied by Nazi Germany to have fewer Denisovans than countries that were not occupied by Nazi Germany. This is exactly what you find, in that Belarus and Poland have absolutely no Denisovan people, and Germany has almost none. Germany, Poland, and Belarus contain matches to Ashkenazi Jews (at 90% of the genome), which you can see in the chart below, suggesting that the Denisovan Jews were disproportionately impacted by the Holocaust. That is, even though some people in these countries are genetically Jewish, you don’t find any Denisovans, suggesting that Denisovans generally were nearly annihilated during the Holocaust. Moreover, Finland avoided occupation by both Germans and Russians, and did not actively persecute Jews, and as you can see in the first chart above, they still have a sizable Denisovan population. In contrast, Russia has no relationship to Denisovans, and the Russians also persecuted Jews, separate from the Holocaust. Though we cannot know using this data alone what the populations of Europe looked like prior to the Holocaust, it’s at least consistent with the assumption that Denisovan populations were disproportionately impacted by the Holocaust. This is not surprising, given that many Jews are related to Denisovans. However, Jewish populations seem to persist today genetically in all of these countries, which you can see below. This suggests a disproportionate impact on Denisovans that is independent of being Jewish.
Interestingly, Taiwan and Mongolia also have significant Denisovan populations, which you can see in the first chart above. China has no meaningful Denisovan population, and again, Russia has none at all. This suggests at least the possibility that people of Denisovan ancestry actively fled communist revolutions in China and Russia, heading to Taiwan and Mongolia. Now, it is simply not credible to claim that ethnically Taiwanese and Mongolian people are somehow Jewish, especially not in the numbers implied by the chart above. Moreover, it’s not credible to claim that they knew that they were of Denisovan ancestry either. The more rational explanation is that the persecution of intellectuals disproportionately impacted individuals of Denisovan heritage. One sensible explanation is that Denisovans are simply more intelligent than most people. If this is true, then the average IQ in Sweden and Norway (where there isn’t much of a Denisovan population) should be lower than it is in Finland (where there is a sizable Denisovan population).
It turns out this is true, and drastically so. Moreover, the average IQ’s in Norway and Sweden are almost exactly the same, whereas it’s drastically higher in Finland. See the chart above, where Sweden and Norway are at the bottom, just below the U.S., and Finland is ranked 8th, globally, at the top. Russia is slightly off-screen. You simply cannot explain this differential with economics, political dysfunction, or geography, as all three countries are rich, high-functioning, socialist, capitalist, democracies, that have plainly similar geographies, and very similar demographics, save for Finland’s anomalous Denisovan population. Moreover, the average IQ is also higher in Taiwan than it is in China. This all suggests the possibility that by simply killing intellectuals, the Nazis and Communists accidentally killed off our living ancestors, that are also more intelligent than them. The barbarism of the Nazis also sent a ton of scientists to the U.S., so thanks for that. The image above is courtesy of World Population Review.
I’ve noted before that proofs start to fail when you have an infinite number of summands, e.g., in the case of the sum over all Fibonacci numbers, which produces -1. This is obviously wrong, and implies that algebra fails given an infinite number of terms. This suggests that there is a logical independence between limits, and the infinite case, proper.
That is, e.g., the sum, , does not imply that the number 1 is in fact the sum over the infinite set of such terms. You can easily construct a proof by induction that you can always find a value of that will cause the sum to be arbitrarily close to 1. As a consequence, the sum at infinity cannot be less than 1. An intuitive proof would simply note that all terms present in each finite sum, must be present in the infinite sum, and since the finite sums get arbitrarily close to 1, the infinite sum cannot be less than 1. More formally, assume to the contrary that the sum in the infinite case is . Since there is always a value of that will cause the sum to be arbitrarily close to 1, we can therefore always find a value of that will cause the sum to exceed . Therefore, the sum is not less than 1 in the infinite case. The sum can however be equal to 1, without contradicting such a proof by induction (though it could contradict other fundamental assumptions beyond the scope of this discussion), since that proof requires only that all finite sums are less than, yet arbitrarily close to 1. Interestingly, there is similar logical independence in the case that the sum exceeds 1, since that again, does not contradict the proof by induction that the sum gets arbitrarily close to 1 in all finite cases.
I don’t think this is academic, and instead, I think it’s a potentially deep point about algebra in the infinite case, and non-Turing computing, since the sum in the infinite case is not computable, since all computable functions make use of a finite number of operations. Specifically, if infinite systems really exist in Nature, then there could be a correct answer to whether the sum is actually 1, or greater than 1. This reminds us that all of mathematics is ultimately rooted in reality itself, and if our assumptions are wrong, then our theorems will be physically meaningless. For combinatorics (e.g., graph theory, counting problems, etc.), it’s simply not credible to doubt the assumptions, since they’re plainly physically true. But when you get into this kind of mathematics, it’s not obvious what the right answer is.
I’m in the process of unpacking the history of humanity using my machine learning software, and as part of that process, I decided to take a closer look at Denisovans. Specifically, many modern populations have individuals that are a 70% match to Denisovans. In particular, the Jews and Finns have large populations of people that are a 70% match. Below is a distribution that shows a normalized percentage of each population that is at least a 70% match to the Denisovans. The x-axis shows the acronym for the particular population, and all of the acronyms can be found at the end of my paper, A New Model of Computational Genomics [1]. You can also find all the software you need to run these experiments, in addition to the related technical information on alignment, process, etc., in [1].
The natural question is, are all Denisovans the same? Or do they have a unique history of their own? Denisovan remains are generally found in Asia. However, as you can see above, there are modern populations in Europe, Africa, the Middle East, and Asia, that all contain matches to Denisovans. This suggests at least the possibility, that Denisovans have a unique history, that could predate human language altogether. The process I used to test this question is straightforward: First, I found all genomes that are at least a 70% match to at least one Denisovan genome. Then, I constructed clusters, as a second test, like the one below for the Swedish, effectively counting what percentage of each Denisovan population matched to the Swedish Denisovans. As you can see, the Swedish Denisovans are plainly related to the Norwegian Denisovans (which is not surprising based upon geography), though they’re also related to the Chinese Denisovans. Why? Well, Denisovan fossils are generally found in Asia, so this not surprising either.
This is all very interesting on its own, but what’s far more interesting, is that when I tested the distribution of German Denisovans, they failed to match to any of the actual Denisovan genomes. That is, the German Denisovans (a modern population) failed to match to any of the actual ancient Denisovans in the second test. At first I thought I had made a mistake in the code, but I then isolated the Denisovan row of the dataset that the modern Germans match to, and it’s row 378 in the dataset attached below. However, this Denisovan genome itself does not match to any of the other Denisovan genomes, even at 30% of the genome. This suggests that row 378 of the dataset below, is not Denisovan, and is instead, an otherwise unknown species, that seems to be most related to people in the Jharkhand region of India, based upon the chart below, that shows the distribution of matches at 30% of the genome. Note that all of these genomes are taken from the National Institute of Health Database, and the dataset includes provenance files for all genomes, with links to the NIH Database.
It is of course possible that this genome would map to some other Denisovan genome not included in the dataset. However, I would instead wager that this genome is a very early Neanderthal, since it is a match to some Neanderthals at 30%. I think this find, at a minimum, suggests that archeology is limited in some sense, since it doesn’t look to the genome. As such the label Denisovan is questionable in this case. Moreover, the methods introduced in [1], can predict ethnicity (including archaic humans) with an accuracy of about 80%. As a consequence, it’s at least worth looking into. As noted, all of the code you need to run these experiments are included in [1], save for the additional script attached below. The dataset is also attached below.
I introduced a set of algorithms in my paper, A New Model of Computational Genomics [1], that allows you to predict ethnicity with an accuracy of about 80% using mtDNA alone. See Section 5 of [1]. It follows that mtDNA must contain information about paternal ancestry as well, since ethnicity is a combination of maternal and paternal ancestry. See Section 5 of [1] for an explanation, but for intuition, note that men could e.g., select women that have mtDNA bases in common with them, which would over time cause overlap to grow between maternal and paternal mtDNA through selection, rather than heredity, which is impossible with mtDNA, since it is inherited directly from the mother to the child, with little and possibly no mutation at all.
I’m now in the process of applying these methods generally to uncover the origins of humanity, and I believe, I just solved the problem. To begin, we have to accept the astonishing fact that many living human beings are nearly perfect matches to archaic humans. Specifically, the Roma and Papuans (and some others) are a 95% match to Heidelbergensis, the Finns and some Jews (i.e., both Sephardic and Ashkenazi) are a 70% match to Denisovans, and many populations contain people that are a 95% match to Neanderthals. See [1] generally. These percentages are given by (x) the number of matching bases divided by (y) the full mtDNA genome length (around 17,000 bases), after making use of a simple, global alignment. See Section 1.3 of [1]. Again, because the predictions are so accurate, you simply cannot argue with the methods, as they are plainly more precise than haplogroups, which instead should produce an accuracy around chance, and moreover, generally cross national boundaries (e.g., Sweden and Norway are combined into one haplogroup below). See Section 7.1 of [1], and the map below, courtesy of Wikipedia. That is, the methods in [1] are plainly superior to traditional heredity analysis, since they can predict ethnicity at the national level, distinguishing between, e.g., Swedes, Norwegians, and Finns, despite using only mtDNA, and as a consequence, the heredity analysis should also be superior to haplogroups.
I’ve applied the techniques presented in [1] generally, with the goal of discovering the origins of humanity, and I’ve come to the conclusion that all of us descend from Denisovans. This follows from the simple fact that Neanderthals and Heidelbergensis both have a meaningful relationship to Denisovans, whereas Neanderthals and Heidelbergensis have no real meaningful relationship to each other. This is consistent with the hypothesis that both species descend from Denisovans. See Section 6.1 of [1]. I’ve also managed to assemble a fairly detailed portrait of the migration patterns of human beings globally, which is discussed below.
The Peopling of the Pacific
I recently discovered that the people of Hawaii have only minimal connections to archaic humans. Specifically, for the Hawaiians, at or above 33% of the genome, there is no relationship to the Neanderthals, Heidelbergensis, or Denisovans. Remarkably, the same is true of the Ancient Egyptians. Moreover, the Ancient Egyptians and Hawaiians have 99.7% of their genomes in common, and even at 30% of the genome (where you would expect imprecise matches), they have very similar distributions of matching ethnicities. This suggests the astonishing possibility that the Ancient Egyptians either settled Hawaii, or both the Hawaiians and Ancient Egyptians descend from the same people. I have only two Ancient Egyptian genomes, and one Hawaiian genome, but the distributions are very similar, and so I don’t think you can ignore the possibility that the Ancient Egyptians settled at least parts of the Pacific.
In any case, both populations plainly did not mate with archaic humans, in any appreciable amount, since they have so few bases in common with archaic humans. See Section 5 of [1]. For intuition, again, selection can cause two distinct mtDNA lines to converge into a new third genome, which would cause, e.g., homo sapiens to have many bases in common with archaic humans, which is the case with, e.g., many Finns, that have more bases in common with Denisovans than they should without selection. In this case, as you can see in the chart above, which shows the differences between the Hawaiians and Ancient Egyptians at 30% of the genome, the Hawaiians are closer to the Roma populations (e.g., the Iberian Roma, Russians, and Papuans) than the Ancient Egyptians are. Note that IB stands for Iberian Roma, and all of the applicable acronyms can be found at the end of [1]. The chart above is constructed by fixing a threshold match percentage, in this case 30%, and then calculating a normalized percentage within each population that are a match to e.g., the Ancient Egyptians. So, e.g., if one Norwegian is a 30% match to at least one Ancient Egyptian, then a counter is incremented for the Norwegian population, and this is done for every genome in the dataset. Those counters are then normalized to [0, 1]. The chart above shows the differences between the match distributions for the Ancient Egyptians and Hawaiians, producing a chart over the interval [-1, 1].
One sensible hypothesis is that the Hawaiians mated at least somewhat with the Papuans, causing them to converge slightly to the Roma lineage. They’re also closer to the Javanese and the people of the Solomon Islands than the Ancient Egyptians are. This makes perfect sense, since the people of Hawaii presumably came from somewhere in Asia, initially settled islands closer to Asia (e.g., Java, the Solomon Islands, and Papua), and only eventually spread to the deep Pacific. Keep in mind, the charts above were generated using a 30% match, and as a consequence, this relationship is not very strong, and instead highlights even subtle differences between the Ancient Egyptians and Hawaiians. You’ll also note that the Ancient Egyptians are somewhat closer to the Thai. Putting it all together, one sensible hypothesis is that some Thai people sailed further into the Pacific, mated with people that were already living in Java, the Solomon Islands, and Papua, and eventually formed an isolated and new people in Hawaii.
If this is true, which is consistent with the mtDNA of the Ancient Egyptians and Hawaiians, then the people of Java, the Solomon Islands, and Papua, should all be ancient and possibly archaic people, since they would have already been in the Pacific under this hypothesis. This is consistent with the fact that the Javanese and Solomon Islands people are a 95% match to some Neanderthals, suggesting the astonishing possibility that Neanderthals knew how to sail over large distances. Similarly, the people of Papua are a 96% match to Heidelbergensis, suggesting the more general thesis, that archaic humans knew how to sail. The net picture would be that the Ancient Egyptians (or their close relatives) avoided mating with archaic humans as a general matter, prior to traveling to the Pacific, and then presumably could not avoid doing so once there, eventually settling Hawaii with somewhat more archaic mtDNA than their Egyptian relatives. This is also consistent with the clear preference for avoiding archaic humans in populations such as the Icelandic, Munda, Basque, and Igbo.
The obvious question is, how did these people get to Hawaii? Unlike Papua, Java, and the Solomon Islands, Hawaii is completely isolated, and extremely far from Asia. Moreover, because the Hawaiians have no appreciable relationship to archaic humans, they must be some of the earliest humans. Logic dictates that they were probably the first humans that learned to sail, at least over distances this large, allowing them to completely avoid archaic humans in remote locations like Hawaii. Further, as these are remote islands, that are impossible to get to without a boat, it follows that the original settlers would almost certainly have had sophisticated seafaring abilities, possibly even telescopes. To understand why, just keep in mind human visibility is extremely limited, and if you simply sail out into the open Pacific, you will have no drinking water, other than what you bring with you, and as a result, any navigational errors will quickly lead to death, in just a few days. As a consequence, they could not have simply stumbled upon these islands, and instead, must have known where the islands were in advance. It’s possible they followed migratory birds, but again, birds can travel much faster than a boat, at times, and as such, if you lose the birds, you might again find yourself dead. Moreover, some birds can travel thousands of miles without rest, implying that again, unless you know where the birds are going beforehand, you could end up in the open Pacific, and therefore dead. It is instead more sensible to assume that people capable of building giant pyramids that stand to this day, were also capable of fabricating telescopes, which, because they’re presumably made of glass, and probably small, might not survive thousands of years, possibly longer, depending upon when these people actually showed up.
The Migration-Back Hypothesis
The Icelandic people, who are also geographically isolated, have no relationship with archaic humans at or above 33% of the genome. However, this is not limited to geographically isolated people, specifically, the Basque, Igbo, and Munda people, who are all closely related to each other, and the Thai, have no relationship to archaic humans at or above 33% of the genome. In contrast, the Norwegians have no relationship with archaic humans at or above 96% of the genome, and for all percentages below that, there is a non-zero relationship to Heidelbergensis. Note that this does not mean that all Norwegians are a 96% match to Heidelbergensis, and instead means that at least some Norwegians are a 96% match to Heidelbergensis. This suggests the general premise that some isolated peoples (whether geographically or culturally) have managed to avoid mating with archaic humans. However, Iceland was relatively recently populated by Nordic people around 1,000 AD, and Iceland has no indigenous people. Because the Norwegians are Nordic, just like the Icelandic, and all of these populations apparently avoided archaic humans generally, when compared to others, it follows that the migration to Iceland, by the Nordic people, could have been at least partially motivated by a desire to remain genetically isolated from archaic humans.
As a general matter, Scandinavia presents spectacular evidence for the hypothesis that some Asians migrated back from Asia, to Europe and Africa (i.e., the migration-back hypothesis). Specifically, the Swedes and Igbo are close to the Munda of India, whereas the Norwegians and Nigerians generally, are close to the Thai and the Munda. This is obviously consistent with a migration-back from Asia, in this case, with two distinct groups, making basically the same journey back, splitting into a Northern European group (the Swedes and Icelandic, on one hand, and Norwegians on the other) and an African group (the Igbo and Nigerians generally, respectively).
The obvious question is, how is it that completely morphologically distinct people are all so closely related to each other? In particular, some Norwegians and Nigerians are a 99.7% match, and many are a 99.0% match, and therefore nearly identical on the maternal line. This is completely contrary to common intuition, which is that morphologically distinct people, should have major differences in their genetics. You can argue that because we’re looking only to mtDNA, that the picture is limited, and this is undoubtedly the case. However, as noted, the methods in [1] are able to predict ethnicity with 80% accuracy, and as a consequence, it’s not rational to ignore such a high match count between populations. One sensible hypothesis is that both groups descend from a common set of ancestors, ultimately from Africa, that migrated to Asia, and then migrated back, splitting into two groups, one moving to Scandinavia, the other moving to Nigeria. I would wager that this migration occurred prior to the development of modern human appearance, and that we were anatomically modern, but still perhaps even without complexion altogether, for the simple reason that we might not have lost our body hair. This would over time, allow the two populations to develop distinct appearances, without changing mtDNA at all. Note that mtDNA can remain stable for thousands of years, and as such, the histories we’re considering are in the tens of thousands of years, and possibly longer. For the Norwegians, it would be much easier to avoid mating with other people than it would have been for the Nigerians, for the simple reason that Norway is geographically isolated, but the Basque and Igbo (also from Nigeria) show us that it is possible. Moreover, as noted, the Norwegians do have an appreciable relationship to archaic humans, whereas the Basque and Igbo do not, suggesting that cultural isolation might be a more powerful factor in avoiding archaic humans. In any case, the overall conclusion, is that some Europeans, Africans, and Asians have ancient relationships, that could predate the modern superficial distinctions between human beings, all of which is consistent with a migration-back hypothesis.
The Overall Migration History of Humanity
Putting the peopling of the Pacific in the context of the migration-back hypothesis, it seems likely that Neanderthals and Heidelbergensis had already learned to sail and settled somewhat remote locations like Papua and Java. Sometime afterwards, homo sapiens travelled North East, from Africa to Central Asia, specifically, somewhere near Kazakhstan. See Section 6.1 of [1]. Then, some of those homo sapiens travelled back to Africa (e.g., the Ancient Egyptians), whereas others travelled further East, eventually into the Pacific. This would explain the otherwise inexplicable relationships between e.g., the Ancient Egyptians and Hawaiians, and the Scandinavians, Africans, and Asians generally. That is, the migration-back hypothesis, and the theory of the peopling of the Pacific above, together form a fairly complete portrait of the macroscopic history of humanity.
Who Were the Vikings?
Although it might seem tangential to the bigger picture of history presented above, this exact same analysis, using the same populations, can be applied to the case of the Vikings, revealing a perfectly sensible answer as to who they were, that is consistent with not only genetics, but archeological evidence, historical evidence, linguistic evidence, and common sense. The answer is in my opinion, that they were a subset of the Scandinavian people that lived primarily (at least at some point) in South East Sweden, with ancient connections to the Finns. The basic intuition for this hypothesis follows from the distribution of Rune Stones, about half of which are located in Sweden. Within Scandinavia itself, Sweden has about 2000 Rune Stones, whereas Denmark has about 250, and Norway has 50. You can see in the map below, courtesy of Wikipedia, that the distribution of Rune Stones in Sweden is concentrated in South East Sweden. This is of course close to Finland, and moreover, the genetic evidence I’ll present also suggests an ancient connection to modern day Finns.
As noted above, the Jews (i.e., both Ashkenazi and Sephardic) are also related to the Denisovans. This does not imply that the Vikings were Jews, though you can’t ignore the obvious fact that the Danes, Finns, Irish, and Jews, are all closely related to the Denisovans (see the chart below).
As a matter of religion (as opposed to genetics) modern Finns are generally not Jewish (they are predominantly Christian), and moreover, in the past, they practiced a form of Paganism, not Judaism. That said, the geography of the Rune Stones suggests at least the possibility of a unique people, and moreover, there appears to be a genuine connection between the Canaanite religions and languages, and the Vikings. Specifically, the Vikings had a god named Odin, whose son was Baldr, and the Canaanites had a god named El or Adon, whose son was Baal. Moreover, there are strange similarities between the Phoenician alphabet, the Runic Alphabet, and an Ancient Finnish Alphabet known as Karelian, which is shown below, courtesy of Wikipedia. Finnish is an Uralic language, and it’s certainly not accepted theory that Phoenician is Uralic, though that’s not the point in any case. The point is instead, it seems at least plausible that the Vikings borrowed culture and language from the Middle East.
Finally, there’s at least one example in Viking art, of what might be a Hamsa (the hand with an eye in it, bottom left of center, in the image below), and possibly a Phoenician-style eye (the two eyes, one in the figure’s head, the other external, suggesting a spirt or deity). You can also see the resemblance between Karelian and Phonecian, and the scripts in the image below. That said, the Vikings were extremely well-travelled, and certainly adopted religious symbols from other cultures, in particular, Buddha. As a consequence, I don’t think we can read too much into the art, though the alphabet is plainly reminiscent of Phoenician and Ancient Finnish, which when coupled with the apparent overlap in deities, suggests a bona fide connection to the Middle East, that defined a unique group of people in Scandinavia. The image below is of a Viking artifact found in Funen, Denmark, courtesy of Wikipedia.
All of that said, none of this evidence is as compelling as the genetics itself. Specifically, as noted above, selection by one group with respect to another, can cause the two groups to converge genetically. Despite the fact that a larger portion of the Ashkenazi population is a 70% match to the Denisovans (see the chart above), it turns out that the non-Denisovan Finns are closer to the Denisovans than the non-Denisovan Ashkenazi. That is, if you look at the Finns and the Ashkenazi that are not a 70% match with the Denisovans (i.e., every individual that does not contribute to the chart above), you find that these Finns have more bases in common with the Denisovans than the non-Denisovan Ashkenazi, but the difference is slight, with an average of about 11 more bases. This is consistent with a relationship between Finns and Denisovans that is somewhat more ancient than that which is between the Ashkenazi and the Denisovans. That is, the Denisovans lived in Finland for a very long time, and as a consequence, the mtDNA of Denisovans converged significantly with the local population, and slightly more than that of the Ashkenazi. Counterintuitively, despite the fact that there are fewer living Denisovan matches in Sweden and Norway (again, see the chart above), the match between non-Denisovan Norwegians and Swedes is even stronger than the match with the Finns. This suggests more intense selection for Denisovan mtDNA, causing Norwegians (with 76 more bases in common than the Ashkenazi) and Swedes (173 more bases in common) to be even closer to Denisovans, despite having a much smaller truly Denisovan population than Finland and the Ashkenazi.
This is the intuition for the hypothesis that the Vikings were actually related to ancient Finns, and not Ancient Swedes, despite the location of the Rune Stones in Sweden. Specifically, present-day Finland has the largest percentage-wise population of Denisovans in Scandinavia (see the chart above), and so it is sensible to assume that the Denisovans in the rest of Scandinavia originated in Finland. Moreover, Denisovan remains are generally found in Asia, and not anywhere else. Common sense suggests that Denisovans migrated West from Asia to Finland, and some of them moved on to other areas in Scandinavia and elsewhere (possibly e.g., Estonia, given the language groups). Moreover, there are probably not many living people related to Denisovans in Russia (see the chart above), despite Denisovan remains in Asia generally, suggesting that the Denisovans fled West to Finland, and beyond.
Because the Vikings settled Iceland and Dublin, we should find a similar relationship to the Denisovans there. Specifically, if the Vikings were at least part Denisovan, then we should find Denisovans in Iceland and Ireland, and moreover, among those that are not a 70% match to Denisovan, we should find evidence of selection for Denisovan mtDNA. This is exactly the case, as the Irish have a significant Denisovan population (see the chart above), and moreover, though I have only one Icelandic genome, and one genome from Dublin, they are a 99.7% match to each other. Moreover, both exhibit not only strong selection for Denisovan mtDNA, but the strongest among the Scandinavians (with about 250 more bases in common with Denisovans than Ashkenazi), which is consistent with the hypothesis that the Vikings were related to modern day Finns, and therefore significantly Denisovan.
Finally, there is some genetic evidence that the connections between the Vikings and the Middle East are genetic, and not merely the result of, e.g., trade between the Middle East and the Vikings, which definitely happened. Specifically, the Dublin genome is a 99.87% match to a very large number of Sephardic Jews, and a decent number of Pashtuns. They’re also a match to the Ukrainians, but this is not surprising, given interactions between the Vikings and Ukrainians. Finally, they’re also a decent match to the Swedes, Ashkenazi, Germans, and Scotts, and while you might question the connection to the Ashkenazi, the obvious truth is that Ashkenazi Jews are very close to Northern Europeans generally. Taken as a whole, this is probably the right distribution.
The same is true to a marginally lesser extent of the Icelandic genome, which is a 99.75% match to the same populations, though this genome is a match (albeit at a lower threshold) to more Scandinavians. All of this would make perfect sense, if at least some of the Vikings were Canaanites. Specifically, if they were Phoenician, then this would explain basically everything, including their ability to build ships and sail large distances, and perhaps even the timing. The Phoenicians were conquered by the Romans around 64 BC, and the Vikings came to fruition about 1,000 years later, which leaves plenty of time. Putting it all together, I’d wager that a group of people from the Middle East somehow found their way to Scandinavia, and this set the spark to the flame that became the Vikings, and eventually modern Scandinavia.
The Code and the Dataset
All of the code you need to run these examples is linked to in [1], and the dataset is here.


![[0,1]](https://s0.wp.com/latex.php?latex=%5B0%2C1%5D&bg=ffffff&fg=444444&s=0&c=20201002) , that sum to 1, for the simple reason that the bases must be one of the four. However, note there are minor deviations due to missing bases. The distribution of bases over the entire dataset are unequal, and that’s interesting in and of itself. However, I just tested the distribution of bases within each population ethnicity, to see if there’s meaningful variation in the distribution of bases. It turns out there is meaningful variation, suggesting at least the possibility that different populations select for different distributions of bases.
, that sum to 1, for the simple reason that the bases must be one of the four. However, note there are minor deviations due to missing bases. The distribution of bases over the entire dataset are unequal, and that’s interesting in and of itself. However, I just tested the distribution of bases within each population ethnicity, to see if there’s meaningful variation in the distribution of bases. It turns out there is meaningful variation, suggesting at least the possibility that different populations select for different distributions of bases.


 , does not imply that the number 1 is in fact the sum over the infinite set of such terms. You can easily construct a proof by induction that you can always find a value of
, does not imply that the number 1 is in fact the sum over the infinite set of such terms. You can easily construct a proof by induction that you can always find a value of 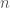 that will cause the sum to be arbitrarily close to 1. As a consequence, the sum at infinity cannot be less than 1. An intuitive proof would simply note that all terms present in each finite sum, must be present in the infinite sum, and since the finite sums get arbitrarily close to 1, the infinite sum cannot be less than 1. More formally, assume to the contrary that the sum in the infinite case is
that will cause the sum to be arbitrarily close to 1. As a consequence, the sum at infinity cannot be less than 1. An intuitive proof would simply note that all terms present in each finite sum, must be present in the infinite sum, and since the finite sums get arbitrarily close to 1, the infinite sum cannot be less than 1. More formally, assume to the contrary that the sum in the infinite case is  . Since there is always a value of
. Since there is always a value of  . Therefore, the sum is not less than 1 in the infinite case. The sum can however be equal to 1, without contradicting such a proof by induction (though it could contradict other fundamental assumptions beyond the scope of this discussion), since that proof requires only that all finite sums are less than, yet arbitrarily close to 1. Interestingly, there is similar logical independence in the case that the sum exceeds 1, since that again, does not contradict the proof by induction that the sum gets arbitrarily close to 1 in all finite cases.
. Therefore, the sum is not less than 1 in the infinite case. The sum can however be equal to 1, without contradicting such a proof by induction (though it could contradict other fundamental assumptions beyond the scope of this discussion), since that proof requires only that all finite sums are less than, yet arbitrarily close to 1. Interestingly, there is similar logical independence in the case that the sum exceeds 1, since that again, does not contradict the proof by induction that the sum gets arbitrarily close to 1 in all finite cases.










