I’m still tweaking this, but this is an algorithm for finding common ancestry given mtDNA (it will not work otherwise), and the best fit for a common ancestor to a population.
https://www.dropbox.com/s/yxvqgt73gfxxqlo/Root_Testing_CMNDLINE.m?dl=0
I’m still tweaking this, but this is an algorithm for finding common ancestry given mtDNA (it will not work otherwise), and the best fit for a common ancestor to a population.
https://www.dropbox.com/s/yxvqgt73gfxxqlo/Root_Testing_CMNDLINE.m?dl=0
I’ve never been able to prove formally why my unsupervised classification algorithm works, and in fact, I’ve only been able to provide a loose intuition, rooted in how I discovered it: as you tighten the focus of a camera lens, the changes near the correct focus are non-linear, in that the object quickly comes into focus. And so I searched for the greatest change in the structure of a dataset as a function of discernment, which works incredibly well, especially for an unsupervised algorithm. In contrast, the supervised version of that algorithm has a simple proof, which you can find in my paper Analyzing Dataset Consistency. However, it just dawned on me, I think I explained why it works, though it’s not a formal proof, in my other paper, Information, Knowledge and Uncertainty. Specifically, the opening example I give is a set of boxes, one of which contains a pebble, where the task is to guess which box the pebble is in. If someone tells you that the pebble is not in the i-th box, then your uncertainty is reduced. But the reason it’s reduced is because the system is now equivalent to a system with one less box. In contrast, the rest of the examples I give in that paper, deal with static observations that have some fixed uncertainty.
Applying this to my unsupervised clustering algorithm, the point at which the entropy changes the most (i.e., Uncertainty), is also the point at which your Knowledge changes the most, due to the simple equation 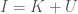
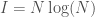
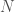
In any case, this is not a proof, but it is a mathematical explanation. What I’m starting to come around to, is the idea that some phenomena, perhaps even some algorithms, function as a consequence of epistemological truths of reality itself. You can definitely accuse me of laziness, in that I can’t formally prove why the algorithm works, but that dismisses the possibility that some things might be true from first principles that defy any further logical justification, in that they form axioms consistent with reality itself. In that case, there is no proof beyond the empirical fact that Knowledge changes sub-optimally past the point identified by the algorithm. The reason I believe this is possible, is because the equation
The reason I thought of this, is because I was working on clustering populations on the basis of mtDNA, and I noticed the same thing happen that happened when I first started my work in A.I. –
There was a massive discontinuous change in cluster entropy, as a function of the inclusion threshold. When I looked at the results, it produced meaningful population clusters, where e.g., both Japanese and Finnish people are treated as homogenous, and basically everyone else is heterogenous. This was totally unsupervised, with no information at all, other than raw mtDNA, and it’s obviously correct. Moreover, Sweden and Norway produced basically the same heritage profile, and even terminate at exactly the same iterator value –
This is consistent with the fact that Norwegians and Swedes are genetically closer to each other than they are to the Finns. The Finns also speak a totally different, Uralic language, whereas Norwegian and Swedish are both Germanic, and so in this case, heritage follows language, which is not necessarily always true, for the simple reason of conquest. For example, the Swedes and Norwegians had their own alphabet, the Runic Scripts, and now they don’t, they use the Latin alphabet like everyone else, because of what is basically conquest.




Above are the plots for the Finnish, Japanese, Swedish, and Norwegian heritage profiles I mentioned, and below is the code and a link to the dataset. You might be wondering how it is that of the few populations that map to Finland, Nigeria is among them. Well, it turns out, 87% of the complete Finnish mtDNA genome maps to a 2,000 year old Ancient Egyptian genome. They also map basically just as closely to modern Egyptians. All of this data is from the National Institute of Health, and you can find all of it by entering the following search query into the NIH Database:
ETHNICITY AND ddbj_embl_genbank[filter] AND txid9606[orgn:noexp] AND complete-genome[title] AND mitochondrion[filter]
Just replace ETHNICITY with Norway, Egypt, etc. Isn’t life something when you actually do the work.
Here’s the code:
https://www.dropbox.com/s/h8ae0tuvtoa1bk1/Percentage_Based_Clustering_CMDNLINE.m?dl=0
Here’s the dataset:
https://www.dropbox.com/s/mj8qk8jxybc9wbc/MTDNA.txt?dl=0
The dataset now includes 19 ethnicities (listed below), and it’s simply fascinating to dig into, and there’s a bunch of software in the previous posts you can use to probe it.
Kazakh, Nepalese, Iberian Roma, Japanese, Italian, Finnish, Hungarian, Norwegian, Sweden, Chinese, Ashkenazi Jewish, German, Indian, Switzerland, Nigerian, Egyptian, Turkish, English, Russian.
I noticed that when you compare a population to itself, at least using mtDNA, it produces a characteristic profile that is unique to the population, as shown in the graph below that plots the results of comparing the mtDNA (full genome) of 10 Swiss people to each other.

Specifically, when counting matching bases between a given row (i.e., individual), and the rest of its population, the average (A), standard deviation (S), minimum (m), and maximum (M) of the number of matching bases, viewed as a vector 

The idea is you do this for every genome in a given population individually, and this will construct a dataset of vectors in the form of P, one for each genome in the population (i.e., a number of rows equal to the number of genomes, each row in the form of P, together forming a matrix with four columns). Note however, you’re comparing genomes to other genomes in the same population (e.g., German mtDNA compared to German mtDNA). You then do this for every population, separately, constructing unique matrices for each population (i.e., one matrix for German, Italian, etc.).
Now combine all of the matrices into a single matrix dataset, and treat the known classifiers as unknown, and try to predict the classifier of a given profile (in this case nationality, e.g., German).
Simply run Nearest Neighbor, mapping 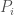
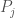
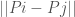
I did exactly this over a dataset of 172 full genomes, from 18 populations, and the accuracy was 82.56%, without any other refinement. The total runtime was just 0.390 seconds, running on a MacBook Air. There is no way you’ll achieve this kind of runtime using Neural Networks. Each of the populations has only 10 full genomes, suggesting higher accuracy could be possible by simply increasing the size of the dataset. Other techniques can likely also improve accuracy.
Because mtDNA is inherited directly from the maternal line, if it weren’t for mutations, a perfect copy would be passed on from mother to daughter, etc, with the male line’s mtDNA simply vanishing. Nonetheless, we know that there is significant variation in mtDNA, which implies significant mutation. However, this work shows unambiguously that there is local variation, in that people that occupy the same present geographies, have similar mtDNA. This simply doesn’t make sense, unless there is some environmental impact on the mutations on mtDNA, causing people in similar environments to have similar mutations.
I think instead, it’s far more reasonable to assume that the male line actually does impact mtDNA indirectly, through the genetic machinery, which must be at least partly inherited from the paternal line. That is, the mechanisms that read and replicate DNA, and produce proteins, are not to my knowledge inherited from either sex exclusively. This implies that common paternal lines could produce similar mutations, which would explain the local similarity of mtDNA, and still allow for mutation. This implies that the paternal line could be discoverable through mtDNA alone, through the analysis of similar mutations on the same maternal line. For the same reasons, it implies that people with highly similar mtDNA would have similar paternal and maternal lines.
Here’s the dataset:
https://www.dropbox.com/s/mj8qk8jxybc9wbc/MTDNA.txt?dl=0
Here’s the code:
https://www.dropbox.com/s/y59ezd50k8df2sa/Build_Class_Profiles_CMNDLINE.m?dl=0
https://www.dropbox.com/s/e07o4x9fhlwxo2d/NN_fully_vectorized_BlackTree.m?dl=0
https://www.dropbox.com/s/b5d19n63q9q85zh/Genetic_Nearest_Neighbor_Single_Row.m?dl=0
THIS WAS THE RESULT OF BAD DATA, DISREGARD. THE ALGORITHMS ARE HOWEVER GOOD IN CONCEPT.
I’ve assembled a dataset using complete mtDNA genomes from the NIH, for 10 individuals that are Kazakh, Nepalese, Iberian Roma, Japanese, and Italian, for a total of 50 complete mtDNA genomes. Using Nearest Neighbor alone on the raw sequence data, the accuracy is about 80%, and basic filtering by simply counting the number of matching bases brings the accuracy up to 100%. This is empirical evidence for the claim that heritage can be predicted using mtDNA alone. One interesting result, that could simply be bad data, the Japanese population (classifier 4 in the dataset), contains three anomalous genomes, that have an extremely low number of matching bases with their Nearest Neighbors. However, what’s truly bizarre, is that whether or not you include these individuals in the dataset (the attached code contains a segment that removes them), generating clusters using matching bases suggests an affinity between Japanese and Italian mtDNA. This could be known, but this struck me as very strange. Note that because matching bases is plainly indicative of common heritage, this simply cannot be dismissed.


The chart on the left shows accuracy as a function of confidence, which in this case is simply the number of matching bases between an input and its Nearest Neighbor. Note the x-axis on the left does not show the number of matching bases, and instead shows the ordinal index of the number of matches (i.e., a x value of 25 is the maximum number of matching bases, which is approximately 17,000). The chart on the right shows the distribution of classes in the clusters for the Japanese genomes, after removing the three anomalous genomes. Clusters are generated by fixing a minimum number of matching bases, in this case it’s fixed to the minimum match count for all Japanese genomes and their respective Nearest Neighbors. Any other genome that meets or exceeds this minimum is then included in the cluster for a given genome. Note the totals can exceed the size of the dataset, since the clusters are not mutually exclusive, and so e.g., the clusters for two Japanese genomes can overlap, adding to the total count using the same genomes. As you can see, it shows a strong affinity between Japanese and Italian mtDNA. The analogous chart for the Italian population shows a similar affinity for Japanese mtDNA. No other groups show any comparable affinity for Japanese mtDNA. Because DNA is finite, and e.g., mtDNA has a well-defined sequence length, the number of possible sequences is fixed. As a consequence, as you increase the minimum number of matching bases required for inclusion in a cluster, the number of possible sequences that satisfy that minimum decreases exponentially as a function of the minimum. Therefore, if you increase that minimum, groups that do not actually belong should drop off exponentially. Those that remain, at a rate that is not exponentially decaying are more likely to be bona fide members of the cluster.
Here’s the dataset, which you can expand upon by entering the following Query into the NIH Database:
“ddbj_embl_genbank[filter] AND txid9606[orgn:noexp] AND complete-genome[title] AND mitochondrion[filter]”
Here’s the code:
Attached is code that implements an analog of my core supervised classification algorithm to genetic sequences. The original algorithm operates on Euclidean data, and because genetic sequences are discrete, this algorithm simply increases the minimum matching number of bases between two sequences, rather than increasing a spherical volume of Euclidean space. You can read about the original algorithm in my paper, Vectorized Deep Learning, and this works exactly the same way, it’s just not Euclidean. I tested it on three datasets from Kaggle, that contains raw genetic sequences for dogs, humans, and chimpanzees, the classification task being to identify common ancestor classes, separately, for each species (i.e., it’s three independent datasets). The accuracy was roughly 100% for all three datasets. The runtimes were 78 seconds (695 training rows, 122 testing rows, 18,907 columns); 371.314 seconds (1,424 training rows, 251 testing rows, 18,922 columns); and 1,752 seconds (3,085 training rows, 544 testing rows, 18,922 columns), respectively. This is consistent with how this algorithm performs generally, as it is extremely efficient and highly accurate. The only difference here is that the data is not Euclidean. This is more empirical evidence for the claim that genetic data is locally consistent, which in turn implies that polynomial-time algorithms can be used to accurately classify genetic data.
The Shannon Entropy is not a good measure of structural randomness, for the simple reason that all uniform distributions maximize the Shannon Entropy. As a consequence, e.g., a sequence of alternating heads and tails 
However, Kolmogorov Randomness is too strong of a requirement, since there are sequences that are intuitively random, that are not Kolmogorov Random. Specifically, consider e.g., a Kolmogorov Random string 

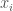
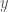
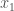
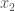


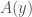

This suggests a simple and intuitive definition of a random string, where a string 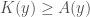
This is quite nice, because it’s simple, and it allows for sequences that have arbitrarily long periods of stability to still be treated as random. And as a consequence, if a sequence has an extremely low entropy, it could still be random in this view, if its subsequences cannot be compressed beyond identifying their lengths.
I published some code that iteratively applies Nearest Neighbor until you create a loop. That is, you run Nearest Neighbor on its on output until you return the same row twice. Symbolically, if 


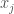
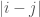
But it just dawned on me, if you don’t allow it to create a loop, and instead delete any row that produces a loop, and apply it again, until you find a unique output, it will force you to iterate through the entire dataset, quantizing the distance between a given starting row, and all other rows. If you do this for a population of genetic sequences, e.g., using human genomes, you can then define the distance between a member of a given population, and all other populations, globally. If you do this repeatedly, you will create a distribution of distances from one population to all others. So if for example you start with a large group of Chinese people, and then apply Nearest Neighbor, using a subset of the entire world population as your dataset (i.e., all major geographies), you will construct a quantized distribution of distances from Chinese people to all other people, which will of course probably iterate through a large number of Chinese people before moving on, but it will eventually move on.
This distribution will also of course create averages, allowing you to say that, e.g., on average Chinese people are a quantized distance of one, two, etc., from populations A,B, and so on. It will also allow you to identify anomalies, since everyone will fall within that distribution, at some distances from the averages. This is now doable, because of the IGSR Dataset, and I’m just starting to download files, which are extremely large, so this will take some time.
If you can figure out a mapping across species, you could in theory apply this process to the entire set of known organisms, creating a distance metric that covers all species.
Fields are plainly a source of free energy, in that e.g., gravity can cause arbitrary blue shifting in photons, and arbitrary acceleration in masses. You can fuss about Potential Energy, but it’s nonsense, especially in the case of a photon, that literally gains Kinetic Energy that was not there before through blue shifting. The more sensible theory of Potential Energy, is that when it is present in a system, it is actually Kinetic Energy that causes no macroscopic motion, and instead causes only small scale motions that must on average “net out” to nothing at the macroscopic level.
Biological life plainly makes use of fields, beyond the obvious of our nervous system, and instead it is a fundamental part of the storage and use of energy in even bacteria, where small scale differences in charges drive molecular machines that, e.g., allow for the production of ATP:
As a consequence, if it’s possible to extract more energy through fields than is consumed through e.g., combustion, then life itself should have figured out how to do this by now, since there is to our knowledge, no laboratory that’s been around for longer than life itself. The general idea being that because it would be advantageous to extract more energy from a field than is consumed in accessing the field, and because the Earth is so large and diverse, and old, it should have occurred by now. This should be measurable, but it would likely require careful, comprehensive, and invasive measurement of all energy consumed by and produced in an organism.
Note that this does not imply perpetual energy, and instead implies that a small bang (from e.g., combustion) is then followed by a bigger bang (from e.g., an electrostatic field), causing the entire process to have a positive yield. This second production of energy might not be translatable back into e.g., chemical energy, and so no infinite loop is necessarily produced by such a process. That said, if it actually turns out that some life generates more energy than it consumes, then it is obviously worthy of study, since human beings might able to adapt these processes into energy producing systems, that would by definition be more efficient than e.g., combustion alone. The general mechanical intuition being that you use chemical energy, or mechanical energy, to access the free energy of some field, likely magnetic, or electrostatic, but anything is possible, what works is what matters.
I’ve posited that anti-gravity is the force that keeps one moment in time from intruding into another, and this makes perfect sense, since it completes the symmetry of gravity, and would force integrity on time itself, preventing the future, past, and present from interacting, as a general matter, though it’s obviously a highly speculative idea. I’ve also speculated that it’s not perfect, which would allow for small scale, spontaneous particles and energy to appear without any ultimate cause, with some low probability, which from what I remember, is what actually happens.
The other day I realized that this could be used to explain Quantum Interference, though I’ll concede at this point, I’ve introduced a number of theories for this phenomenon. The basic idea is that a particle literally interferes with its most proximate moment in the future, causing it to behave as if there were a multitude of instances of itself. In this view, this should not happen as often as particles get larger, and that is plainly true at our scale of existence, where this literally never happens.
However, there’s one problem with this theory, which is that the introduction of e.g., an intervening particle destroys the interference pattern generated by the Double Slit Experiment. However, we can remedy this by observing that exchanges of momentum plainly do not cause this to happen, because e.g., the walls that define the experiment are plainly in the scope of the path of the particle, yet their presence does not destroy the interference pattern. As a consequence, we can posit that exchanges of momentum alone that do not change the energy of a particle, do not destroy the interference pattern, and therefore do not change the path of the particle through time, allowing it to still interact with future instances of itself.
We can then also posit that a change in energy (e.g., one caused by the introduction of an intervening particle that collides with the original particle), does change the path of the particle through time, thereby preventing it from interacting with what were otherwise future instances of itself.
The question still remains, what can cause something to transition from a point particle, to a wave? I haven’t really studied physics in years, but I was pretty thorough when I did, and I don’t recall seeing this question answered. So this is no worse off than a Copenhagen interpretation that posits what is basically the diffusion of the energy of a particle over multiple possibilities in one moment in time, and this was in fact my original theory:
https://www.researchgate.net/publication/344047683_A_Combinatorial_Model_of_Physics
The difference here is that we should be able to test whether or not there is in fact unexplainable interference happening generally, at extremely small scales, and I believe this has in fact been observed. Such an occurrence has nothing to do with Quantum Interference, and instead requires a totally separate assumption, and I guess you could invoke the Uncertainty Principle. Though if both explain the same set of observations, and one is universal, requiring a single assumption, instead of two, then it’s a stronger axiom.
One final note, this implies that true acceleration, i.e., a change in energy, not momentum alone (e.g., simply turning the wheel without pressing the gas), does not change the path through time, suggesting that time itself contemplates or connects possibilities that are connected through a single level of energy.
When you have a known set or population generally, with some known measurable traits, it’s a natural tendency to attribute the properties of that set, to new observations that qualify for inclusion in that set. In some contexts, this is deductively sound, and is not subject to uncertainty at all. For example, we know that the set of prime numbers have no divisors other than themselves and 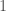

Put all of that aside for a moment, and let’s posit some function that allows you to generate a set, given a single element. If that function truly defines and generates a unique set, then applying that function to the elements of the generated set, should not produce new elements, and should instead produce exactly the same set. Said otherwise, it shouldn’t matter what element I start with, if our function defines a unique set. To create a practical example, view the set in question as a cluster taken from a dataset. This is quite literally a subset of the dataset. There must be in this hypothetical a function that determines whether or not an element of the dataset is in a given cluster. Let’s call that function 
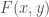
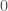


For each such cluster, count the number of elements that are included in the original cluster associated with
We can make this rigorous, using the work I presented in, Information, Knowledge, and Uncertainty [1]. Specifically, your intuitive uncertainty in the attribution of properties of a set to new observations that qualify for inclusion in the set, increases as a function of the number of outbound edges. Similarly, your intuitive uncertainty decreases as a function of the number of mutually connective edges. We can measure uncertainty formally if we can express this in terms of the Shannon Entropy. As such, assign one color to the edges that are mutually connective, and assign a unique color for every other remaining vertex (i.e., element of the set). So if an element 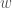
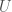


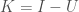
One interesting note, that comes up every time I’ve worked on epistemology, is that if these results are empirically true (and as it turns out the results in [1] are in fact empirically true), it implies that our theory of knowledge itself is subject to improvement, separate and apart from the knowledge we obtain from experiment and deduction. This branch of what I suppose is philosophy therefore quantifies the knowledge that we obtain from empirical results. This study seems similar to physics, in that the results are axiomatic, and then empirically tested. In contrast, mathematical knowledge is not subject to uncertainty at all. And as a result, this work suggests that empiricism requires the careful and empirical study of knowledge itself, separate and apart from any individual discipline. Said otherwise, this work suggests, that empiricism is itself a science, subject to testing and improvement.
